Annals of Neurosciences, Volume 18, Issue 3 (July), 2011
Hallervorden-Spatz syndrome: a rare cause of extrapyramidal manifestations
Corresponding Author:
KV Vinod, MD
Dept. of General Medicine, Jawaharlal
Institute of Postgraduate
Medical Education and Research,
Dhanvantarinagar,Puducherry- 605 006,
INDIA E mail: drkvv@rediffmail.com
Ph.:+91-9442604554,
ABSTRACT
Hallervorden-Spatz syndrome is a rare neurodegenerative disease of autosomal recessive inheritance which presents in childhood or early adulthood with dystonia, dysarthria, rigidity and choreoathetosis. Here we present an unusual case of atypical Hallervorden-Spatz syndrome with onset during adolescence and rapid progression in a young female patient who showed the characteristic “eye of the tiger” appearance on magnetic resonance imaging [MRI] of brain. This reporting intends to highlight Hallervorden-Spatz syndrome as a rare cause of extrapyramidal manifestations and the interesting radiologic picture of the disease.
KEY WORDS: extrapyramidal,pantothenate kinase 2, dystonia, neurodegeneration, eye of the tiger
Introduction:
Hallervorden-Spatz syndrome [HSS] is a rare neurodegenerative disorder of autosomal recessive inheritance characterized by accumulation of iron in basal ganglia. The syndrome encompasses a spectrum of clinically heterogeneous disorders characterized by common features of neurodegeneration and brain iron accumulation. The disease may present in childhood and progress relentlessly culminating in early death [classical HSS] or may present in second or third decade with slow progression [atypical HSS].1 The predominant neurologic manifestations are extrapyramidal and include dystonia, dysarthria, rigidity and choreoathetosis. Involvement of corticospinal tract with spasticity and extensor plantars, cognitive decline, speech disturbances, degenerative retinopathy with visual disturbances and psychiatric disturbances may also be seen.1 Many patients with HSS have mutations in the gene encoding pantothenate kinase 2 (PANK2)2 on chromosome 20p13 and hence this disease is also termed pantothenate kinase associated neurodegeneration. Pathological findings in brain include rust brown pigmentation due to iron deposition, axonal swellings or spheroids and neuronal loss with gliosis predominantly in the globus pallidus and pars reticularis of the substantia nigra.3 This disease is of historical interest and the eponymous term “Hallervorden–Spatz syndrome” has gone into disrepute in view of the unethical activities of the German pathologists Hallervorden and Spatz during the second world war.4 Here we present an unusual case of atypical HSS with onset during adolescence and rapid progression.
Case Report:
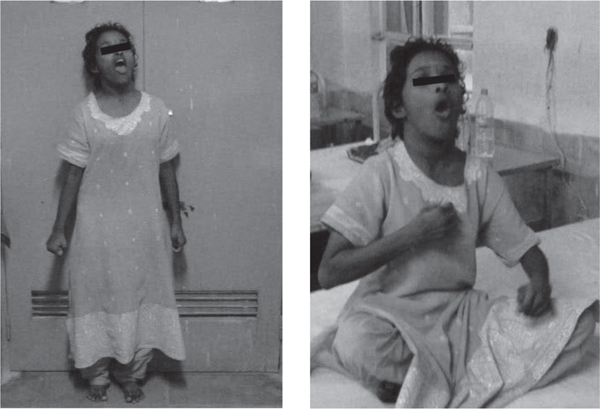
A young girl of 18 years [Figure 1] was brought with progressive complaints of difficulty in walking and abnormal posturing of head and limbs for 4 years. She was apparently well till the age of 14 years when she started having stiffness and abnormal posturing of left leg which made her walk on her left forefoot. The right lower limb was involved in a similar fashion over next 6 months. Patient later developed abnormal twisting movements and awkward posturing of both upper limbs [18 months before current admission]. She was also noticed to keep her head persistently extended with mouth wide open and trunk arched backwards in an awkward manner simultaneously. She had difficulty in speaking and had turned mute for the previous one year. All the abnormal movements subsided during sleep. There was progressive difficulty in walking with frequent falls for the past 1 year but she could still ambulate with assistance. She had stopped attending the school for the past 2 years and till that time her scholastic performance had apparently remained normal. There was no history of seizures, jaundice, visual or psychiatric disturbances. Her birth was uneventful and there was no delay in attaining the milestones. Family history was not significant. Physical examination revealed an average built young female with hypertrophy of both sternocleidomastoids. There was rigidity of all four extremities with cogwheeling and with normal tendon jerks. Both plantars were extensor. She exhibited dystonic posturing of extremities, trunk, head and neck as well as oromandibular dystonia. No tremors or choreoathetotic movements were seen. Slit lamp examination of the eye did not reveal Kayser-Fleischer ring. Fundus examination was unremarkable. Other nervous system and systemic examination was unremarkable. Routine laboratory evaluation was normal. Serum caeruloplasmin level [20mg/dl], 24 hour urinary copper excretion [26µg], serum ferritin [34µg/l] and fasting lipid profile were normal. Peripheral blood film did not reveal acanthocytes. MRI of the brain revealed area of hyperintensity within a region of hypointensity in the medial globus pallidus bilaterally on T2 weighted images-the “eye of the tiger” pattern [Figure 2]. Based on the clinical presentation and the characteristic MRI scan findings, a diagnosis of Hallervorden-Spatz syndrome was made. The patient was given symptomatic treatment with levodopa/ carbidopa combination and later trihexyphenidyl and baclofen were also added. At 2 months follow up, patient has shown only modest improvement of rigidity and dystonia without any improvement in speech.
Fig. 1: showing dystonia of the upper limbs and oromandibular dystonia
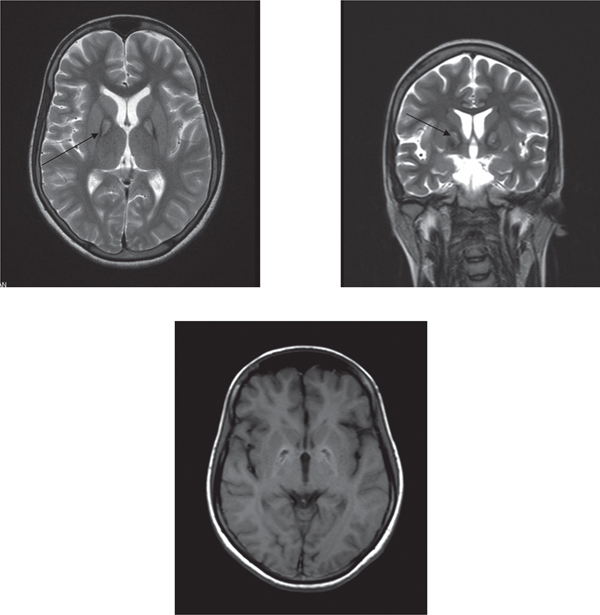
Figure 2: MRI of brain showing area of hyperintensity within a region of hypointensity in the medial globus pallidus bilaterally on T2 weighted images -the “eye of the tiger” pattern [arrows]
Discussion:
Hallervorden Spatz syndrome [HSS] is a rare disease included under the broad category -‘neurodegeneration with brain iron accumulation’ along with acaeruloplasminemia and neuroferritinopathy. The latter two may have similar neurologic features with HSS but can be distinguished from HSS by later onset during adulthood, MRI findings, laboratory tests [undetectable serum caeruloplasmin and low serum ferritin respectively] and genetic testing.1
Our patient had onset of extrapyramidal symptoms like dystonia and rigidity during second decade which were progressive. Patient showed evidence of extrapyramidal and corticospinal tract involvement. Although there was no family history, the above clinical features along with the classical MRI brain picture [“eye of the tiger” sign] and negative tests for Wilson’s disease were sufficient to make a diagnosis of atypical HSS as per the reported criteria.1,5 Sporadic occurrence of the disease as in our patient has also been r eported.5
The characteristic MRI brain pattern of HSS showing area of hyperintensity within a region of hypointensity in the medial globus pallidus bilaterally on T2 weighted images [the “eye of the tiger” pattern] corresponds to the pathological findings. The hypointensity on T2 weighted image is because of iron deposition and central hyperintensity is secondary to gliosis and spongiosis.6 A subset of patients of atypical HSS may not show this radiologic pattern.1
The pathogenesis of HSS involves pantothenate kinase 2 [PANK2] gene mutation2 causing defective phosphorylation of pantothenic acid [required for synthesis of coenzyme A]. This has been postulated to result in underutilization and accumulation of cysteine in basal ganglia which, when in excess causes chelation of iron leading to free toxic radicals production. The preferential involvement of basal ganglia is attributed to the excess of pantothenate kinase receptors. Although genetic analysis to detect PANK2 gene mutation could not be done in our case, the classic MRI pattern of “eye of the tiger” as seen in the index case has been reported to have one to one (strong) association with this mutation.1
In a study of 16 HSS patients reported from northern India,7 the commonest clinical presentation was with limb or cranial onset progressive dystonia as seen in the index case. Other reported manifestations1,5,7,8 like seizures, psychiatric disturbances and acanthocytes in the peripheral blood smear were not seen in our patient. Although, an electroretinographic evaluation could not be done, there was no clinical evidence of optic atrophy or pigmentary degeneration of retina in our patient. Our patient had hypertrophied sternomastoid muscles because of persistent cervical dystonia. She had turned mute because of severe oromandibular dystonia after initial dysarthria. Speech disturbances like palilalia, dysarthria have been described in HSS.1,7 The progression of atypical HSS to loss of independent ambulation is reported to be slower[over 15-40 years from disease onset]1 but rapid progression [within 4 years] as seen in our patient has been rarely described.9
Treatment of HSS is unsatisfactory. It involves pharmacologic [levodopa, trihexyphenidyl, baclofen and bromocriptine] and surgical [deep brain stimulation] interventions aimed at palliation of symptoms.5,9 Botulinum toxin injection has been useful in severe facial and orobuccolingual dystonia to improve speech. Iron chelation therapy has not shown benefit.
References:
1. Hayflick SJ, Westaway SK, Levinson B, et al. Genetic, clinical, and radiographic delineation of Hallervorden-Spatz syndrome. N Engl J Med. 2003; 348: 33–40
2. Zhou B, Westaway SK, Levinson B, et al. A novel pantothenate kinase gene (PANK2) is defective in Hallervorden-Spatz syndrome. Nat Genet. 2001; 28: 345–9
3. Dooling EC, Schoene WC, Richardson EP Jr. Hallervorden-Spatz syndrome. Arch Neurol. 1974; 30: 70–83
4. Shevell M. Racial hygiene, active euthanasia and Julius Hallervorden. Neurology. 1992; 42: 2214–19
5. Swaiman KF. Hallervorden-Spatz syndrome. Paediatr Neurol. 2001; 25: 102–8
6. Adams RJ, Nichols FT, McKie V, et al. Hallervorden spatz syndrome: Clinical and magnetic resonance imaging correlations: Ann Neurol. 1988; 24: 692-4
7. Sachin S, Goyal V, Singh S, et al. Clinical spectrum of Hallervorden-Spatz syndrome in India. J Clin Neurosci. 2009; 16: 253–8
8. Bindu PS, Desai S, Shehanaz KE, et al. Clinical heterogeneity in Hallervorden- Spatz syndrome: A clinicoradiological study in 13 patients from south India. Brain Dev. 2006; 28: 343–7
9. Gregory A, Polster BJ, Hayflick SJ. Clinical and genetic delineation of neurodegeneration with brain iron accumulation. J Med Genet. 2009; 46: 73–80