Annals of Neurosciences, Volume 19, Issue 3 (July), 2012
Neuropathic pain: role of inflammation, immune response, and ion channel activity in central injury mechanisms
ABSTRACT
Neuropathic pain (NP) is a significant and disabling clinical problem with very few therapeutic treatment options available. A major priority is to identify the molecular mechanisms responsible for NP. Although many seemingly relevant pathways have been identified, more research is needed before effective clinical interventions can be produced. Initial insults to the nervous system, such as spinal cord injury (SCI), are often compounded by secondary mechanisms such as inflammation, the immune response, and the changing expression of receptors and ion channels. The consequences of these secondary effects myriad and compound those elicited by the primary injury. Chronic NP syndromes following SCI can greatly complicate the clinical treatment of the primary injury and result in high comorbidity. In this review, we will describe physiological outcomes associated with SCI along with some of the mechanisms known to contribute to chronic NP development.
KEYWORDS: MMPs, Nitric Oxide, TRPV-1, NKCC-1, Cannabinoid Receptors, CB1/CB2, Microglia
Corresponding Author: Daniel K Resnick, MD, MS, Tel: (608) 255-4223, E-mail: resnick@neurosurg.wic.edu
doi : 10.5214/ans.0972.7531.190309
Introduction
Spinal Cord Injury and Neuropathic Pain
Spinal cord injury (SCI) often results in devastating motor and sensory deficits for which current therapy is largely ineffective. Additionally, SCI can induce the development of chronic neuropathic pain states and significantly worsen the quality of life of these patients. A lack of sufficient understanding of the mechanisms underlying NP has affected the development of effective analgesic and restorative therapies. Two of the most common clinical pain behaviors associated with NP syndrome are allodynia and hyperalgesia. Allodynia occurs when normally non-noxious stimuli produce pain and hyperalgesia is the condition of an exaggerated pain response produced by a normally mildly noxious stimulus. The development of some degree of central NP is believed to occur in up to 70% of SCI patients and causes significant discomfort and disability in many areas of a patient’s life.1 It is estimated that up to 1 percent of the population suffers from some degree of NP.2 SCI can produce marked changes in the synaptic circuits of the dorsal horn cells as well as in areas well rostral to the site of trauma through a variety of mechanisms.3 Of particular interest are changes in receptor and ion channel expression and activity, release of local inflammatory cytokines and reactive oxygen species, activation of the immune response in microglia and other immune cells, and the activation of specific intracellular cascades. These are some of the most commonly studied mediating factors that are known to be involved in NP following SCI.
Spinal Cord Contusion Models
A variety of animal models have been designed to study the development of NP following SCI. Some of these models include spinal cord contusion, spinal hemisection, spinal ischemic injury, quisqualte-induced excitotoxic lesions, clip-compression lesions, and argon laser induced lesions.4–6 These models attempts to produce NP symptoms that mimic those observed in a clinical setting, however, the more specific the lesion induced the less clinically relevant the results. Here we examine one of the most commonly employed models used to study central NP development, the contusive spinal cord injury model (cSCI). This is performed using a weight-drop impactor following a laminectomy that spares the dura mater.7 Following the procedure motor deficits and pain behaviors are measured over a set period of time. Common assessments include an open field locomotor test measuring hind limb performance8, a sciatic nerve function index measuring the various relationships between the toes and feet of the hind limbs,9 walking track evaluations10 and an extensor postural thrust measurement which measures the force generated by the hind limbs.11 Changes in response to sensory stimuli are also and important measurement taken following spinal cord contusion.12 Thermal hyperalgesia is typically exhibited beginning about 21 days following injury. Assessments used to quantify the response to this type of noxious heat stimuli are hind paw withdraw latencies and heat threshold tests.13
Inflammatory Response
The inflammatory process is of great significance in the development of NP. Not only does inflammation produce a variety of changes in the extracellular environment, but it also induces profound intracellular changes. Some of the notable components of the inflammation process known to contribute to the manifestation of NP include the accumulation and recruitment of inflammatory cytokines, chemokines and prostaglandins, the modulation of extracellular proteins, changes in transmembrane receptor expression, immune cell infiltration, and intracellular changes modulated via ion channel activity and receptor signaling.14–17 Although the inflammatory response is observed within most tissues in the body following insult, the central nervous system is unique in several important ways. Several distinct cell types are found only in the central nervous system: neurons, astrocytes, oligodendrocytes, and microglia. Further, neurons are known to be limited in the ability to regenerate. The CNS also lacks a lymphatic drainage system and is therefore relatively limited in its ability to expand because of the presence of surrounding tissues such as the dura, spinal canal, and skull. Thus relatively small levels of swelling can have significant consequences. The blood-brain barrier also has a role in producing an inflammatory response unique to the CNS. Endogenous neurochemical mediators and growth factors produced secondary to the injury contribute to trauma induced BSCB disruption, edema, and the subsequently observed motor deficits and NP.18
For our purposes in understanding the development of NP, this complex defense mechanism of inflammation can be separated into two classes: beneficial and detrimental. Inflammation in the CNS serves a function of eliminating pathogens, phagocytizing necrotic tissue, mediating repair mechanisms of some types of damaged tissues, and recruiting leukocytes to the damages area. Additionally, neurotrophic factors released by microglia and astrocytes in response to cytotoxic factors produced by damaged cells have been shown to have some neuroprotective effects.19,20 The inflammatory response, however, is also known to play a significant role in the development of NP syndromes. Many of the same neurotrophic factors that have shown neuroprotective effects, such as BNDF and TNF-α, also enhance the inflammatory response. In doing so, further damage is caused to injured and non-injured tissue alike and is often more severe than the damage caused by the primary insult. Mediators of this secondary damage are due to acute increases in proteases, nitric oxide, bradykinins, prostaglandins, and tumor necrosis factor alpha (TNF-α), among others. In addition, these extracellular factors influence a number of intracellular changes that further promote the development of NP and will be discussed below.
MMPs
Matrix metalloproteases (MMPs) are a family of zinc-dependent extracellular proteases that are involved in the digestion of extracellular matrix components as well as some cell surface proteins like adhesion molecules, receptors, growth factors and cytokines.21 MMP-9 has constitutive expression in uninjured states and is thought to mediate the cellular extracellular environment by assisting in growth and regenerative processes as it is expressed in migrating growth cones.22,23 In injured tissue, MMP-2 and MMP-9 play a key role in the inflammatory response by modulating the development of neuropathic pain. Reactive oxygen species, mechanical stimuli, and mitogen activated protein kinase (MAPK) pathways activate transcription factors such as activator protein 1 (AP-1) and nuclear factor –kB (NF-kB).24 These factors cause a direct increase in MMPs as well as the other powerful inflammatory mediators such as IL-1β and TNF-α. MMPs are thus overexpressed in injured spinal cord tissue and both are thought to mediate NP through IL-1β cleavage. One important difference between the aforementioned MMPs, however, is that MMP-9 and MMP-2 upregulation is correlated with early and later phases of NP respectively.25
Another notable difference between these two MMP profiles is that NP associated with MMP-9 expression is generally associated with microglia activation while MMP-2 related NP is seems to be maintained by astrocyte activation.26
A significant detrimental effect of the overexpression of MMP-9 following SCI is the disruption of the blood-spinal cord barrier. This disruption permits the infiltration of immune cells, such as neutrophils, and inflammatory components which contribute to secondary tissue damage and will interact with resident astrocytes and microglia, subsequently producing even more inflammatory cytokines at the injury site.27 Invading neutrophils will facilitate lesion expansion via necrotic mechanisms and also through the production of reactive oxygen species, proteases, and nitric oxide. Delayed upregualtion of MMP-2 is observed beginning 5 days following cSCI and peaks 7-14 days post-injury.28 Less is known about the expression patterns of MMP-2 following injury, however, it appears that macrophages infiltrating the injury site and astrocytes surrounding the lesion contribute substantially to the induction of MMP-2.29 Knockout studies of MMP-9 deficient mice have demonstrated improved blood-spinal cord barrier integrity following SCI producing diminished neutrophil and macrophage infiltration.30 Treatment with atorvastatin, an MMP inhibitor, post-SCI produced similar results.31 This suggests that inhibition of MMP-9 has a neuroprotective role in following SCI. MMP activity is also implicated in glial scarring following SCI. While this scarring can help to reestablish the blood-spinal cord barrier following injury, the cells within the scar prevent neurogenesis by secreting inhibitory molecules.32 MMP-2 knockout mice show higher prevalence of glial scarring following injury suggesting MMP-2 may also have a beneficial role in improving functional recovery and reducing NP following SCI. Directed inhibition of MMPs may therefore offer an efficient and precise target for the treatment of NP at both early and later phases of presentation.
TIMPs
Matrix metalloprotease tissue inhibitor (TIMP) activity is critical in order to maintain a healthy balance of ECM turnover in the healthy state because it prevents rampant breakdown of ECM proteins by constitutively active MMPs.33 The most common mechanism of TIMP inhibition of MMP activity involves binding of the N-terminal amino acid of the TIMP protein and the zinc ion coordinated to the MMP.34 This interaction between TIMPs and the catalytic domain of MMPs results in conformational changes that prevent MMP proteolytic activity.35 In one study, TIMP-1was shown to inhibit MMP-9 activity, suggesting its induction may have a neuroprotective function in CNS insults.36 As mentioned previously, MAPK activity is implicated in MMP regulation. It is also active in the upregulation of TIMP through the stimulation of c-Jun and c-Fos binding domains.37,38 Interestingly, elevated levels of TIMP expression, induced through activation of cannabinoid receptors, was shown to reduce cancer cell invasiveness.39 This suggests that cannabinoids may provide a therapeutic approach to the attenuation of NP development.
CB receptors
The cannabinoid (CB) system has been implicated as playing an important physiological role in analgesia.40,41 The endogenous CB system includes the CB receptors (CB1 and CB2) and natural ligands (endo-CBs) anandamide (AEA) and 2-arachidonylglycerol (2-AG). The cannabinoid receptors CB1 and CB2 are G-protein coupled receptors that have been shown to be upregulated following CNS insults.42 Their activities have also been implicated in the development of NP by promoting a decrease cAMP production, reducing intracellular Ca++ levels and modulating MAPK activity.43 CB receptors are found predominantly in the presynaptic membrane which allows them to effectively inhibit the release of neurotransmitters. In studies where cannabinoid receptor agonists were administered in animal models of NP, TIMP production was induced and a decrease in hyperalgesic behavior was observed.39 CB1 receptor activity is also implicated in the modulation of NP induced by the TRPV1 receptor through PKA activity. CB1 receptors have been observed in the spinal cord, dorsal root ganglia, and peripheral terminals of c-fibers and are usually found co-localized with TRPV1 receptors.44 It is thought that CB1 receptor activity inhibits the Ca++ dependent MAPK signaling that promotes MMP-2 activity and subsequent NP. Additionally, enzymes that regulate the biosynthesis and catabolism of the endo-CBs are emerging as important modulators of CB system activity levels in NP development. One study demonstrated that CB2 receptor-mediated control of NP was IFN-gamma dependent.45 These studies suggest that the CB receptors attenuate behavior associated with NP through multiple mechanisms. However, NP pain attenuated by CB activation appears to be active only after CNS injury and not peripheral nerve injury. This has been demonstrated using CB1 knockout mice although the mechanism for this difference is not well understood.46
Nitric Oxide
One of the most potent inflammatory mediators is nitric oxide (NO). It induces vasodilation, enhances the production of free radicals, and mediates important cellular activities through the induction of cyclic guanosine monophosphate (cGMP). High levels of NO are also neurotoxic.47 Because the half-life of NO is quite short, the level of NO synthase activity is usually the best measurement of NO induced inflammation. Of the three forms of NO synthase, endothelial, neuronal and inducible, inducible nitric oxide synthase (iNOS) is the primary form active during the chronic inflammatory process.48 The primary cytokines responsible for iNOS upregulation are thought to be TNF-α and interleukin-1b (IL-1β), both of which act through the nuclear transcription factor NF-kB and are induced several hours to several days following injury [49,50]. Constitutively expressed forms of NOS, neuronal and endothelial, play a more significant role in the inflammation response immediately following injury as they are released by the damaged cells. Administration of iNOS inhibitors has been shown to improve function and alleviate NP in chronic constrictive models of SCI.51,52 Further, exogenously administered agmatine, a general inhibitor of NOS, has been shown effective at reducing inflammation, hyperalgesia, and allodynia following excitotoxic SCI.53
Interestingly, evidence also exists to suggest that some endogenous NO activity may have neuroprotective effects following SCI.54 Rats treated with a novel NO-releasing gabapentin derivative following injury demonstrated reduction in pain behavior, although the mechanism is unclear.55 The role of NO in NP is complex and the multiple actions mediated by NO must be considered when seeking to develop therapeutic strategies targeting NOs role in NP development.
Prostaglandin
The prostaglandins are another important mediator of the inflammatory response. These lipid compounds are derived from arachidonic acid through the cyclooxygenase (COX) pathway. Two different isoforms of COX exist, COX-1 and COX-2, and both are implicated in pain and inflammation.56 Inducible COX-2 expression is known to be involved in the inflammatory response 2 hours following SCI and inhibition of COX-2 activity has been shown to improve functional recovery.57 This is thought to be due, at least in part, to a reduction in inflammation. COX-2 is also thought to be inducted in the onset of NP following SCI through the production of prostaglandin E2 (PGE2) and the subsequent activation of microglia signaling.58
Bradykinin
Bradykinin (BK), a peptide generated by the kinin system, is vasoactive and known to have hyperalgesic effects thought to be mediated through interaction with the B1 receptor.16 B1 knockout mice show less NP behavior than their wild-type counterparts.59 Interestingly, BK is also thought to have a moderate neuroprotective role through its attenuation of cytokine release from activated microglia.60 The B1 receptor has been shown to sensitize another receptor implicated in NP, the vanilloid receptor 1 (TRPV-1).44 The upregulation of both the B1 and TRPV-1 receptors in rats demonstrating hyperalgesic behavior following contusive SCI has been observed. Additionally, B1 and TRPV-1 antagonists were shown to have antihyperalgesic effects, even reversing TH behavior, following cSCI.61 Interestingly, B1 attenuation was shown to reduce NP behavior only in rats undergoing cSCI, suggesting that the B1 receptor may be uniquely upregulated following SCI.44
Ion Channel Activity
TRPV-1
As mentioned previously, the TRPV-1 ionotropic receptor is implicated in the hyperalgesic response associated with pain. The TRPV-1 receptor is highly expressed in c-fiber sensory neurons and functions in both thermal and mechanical nociception.62 It is also activated by capsaicin and indirectly by lipoxygenase products.63 Following cSCI, TRPV-1 expression is significantly increased in the dorsal horn in animals exhibiting NP behavior.61. Endogenous TRPV1 agonists have been shown to play a role in increasing NKCC1 activity following painful conditions leading to allodynia.64 Further, TRPV-1 antagonists, such as AMG 9810, have notable antihyperalgesic effects in cSCI rat models.65 Interestingly, cooperative interactions of CB1, CC chemokines, and TRPV1 are likely to play a role in SCI induced changes and NP development at brain level.66
NMDA Receptors
Excitatory glutamatergic receptors are implicated in synaptic plasticity and excitotoxicity involved in the development of NP syndromes.67 N-methyl-d-aspartate (NMDA) receptors are permeable to monovalent ions and calcium and blocked by extracellular magnesium. Glycine also functions as a coagonist with glutamate in activating this channel.68 Direct protein kinase A (PKA) mediated phosphorylation of NMDA receptors is implicated in hyperalgesia and allodynia in central but not peripheral NP syndromes.69 In SCI models, administration of an NMDA antagonist, gacyclidine, leads to improved motor function recovery, and reduced astrogliosis.70 Similarily, administration of a different antagonist, MK-801, produces dose dependent reduction in thermal hyperalgesia but slower analgesic effects.71 Another NMDA antagonist, CHF3381, showed reduced hyperalgesia in improving pain in humans.72 Short acting antagonists have also shown to mediate decreased allodynia and central sensitization following CNS injury.73
Unfortunately, attempts at developing an NMDA antagonist that performs an analgesic function in alleviating NP in human subjects has been slow.74 One possible reason for this is the presence of NMDA receptors throughout the CNS, the non-specific activation of which can produces psychotropic effects.75
GABA Receptors
The GABAergic system is a vital component of nociceptive sensory processing and modulation of its function commonly leads to the development of NP states.76 GABA receptors are found in both pre and post synaptic sites and function as ligand-gated chloride channels.77 Normal GABA receptor function is critically dependent on the activity of intracellular Cl- concentration. Both channel types, A and B, have been implicated in NP syndromes and hyperexcitability. Loss of GABA inhibition also can produce NP. The evidence that hypofunction of GABAergic tone contributes to central NP following SCI is well documented by several experiments althought the exact mechanism by which this occurs is unclear.78,79 Pharmacological treatments that enhance GABAergic function attenuates central neuropathic pain behavior and neuronal hyperexcitability following SCI.80 For example, intrathecal administration of GABA attenuates mechanical allodynia and hyperexcitability of spinal dorsal horn neurons following SCI.81 In addition, treatment with bicuculline (a GABAA receptor antagonist) produces neuronal hyperexcitability and pain behavior in normal rats.82
NKCC1 and KCC2 Activity
The maintenance of intracellular Cl- concentration is essential for proper intercellular communication. The inhibitory activity of GABA receptors is critical for normal neural circuitry function and is particularly sensitive to the concentration of intracellular Cl-. Cation-dependent chloride transporters therefore are of vital importance in understanding NP following spinal cord injury.83 Here we identify two of the major Cl- regulatory proteins, the inwardly directed Na+-K+-Cl- cotransporter isoform 1 (NKCC1) and the outwardly directed K+-Cl- contransporter isoform 2 (KCC2), both known to have in a specific role in NP syndromes.84 Early in development, GABA acts primarily as an excitatory neurotransmitter due to high NKCC1 expression and delayed KCC2 expression, resulting in high levels of intracellular Cl- and thus the maintenance of more excitable states upon activation of GABA receptors.85 Mature GABA neurons have a more positive equilibrium potential. It is known that following cerebral ischemia NKCC1 activation leads to astrocyte induced swelling and glutamate release, as well as neuronal Na+, and Cl- influx during acute excitotoxicity. Inhibition of NKCC1 activity also significantly reduced infarct volume and cerebral edema following cerebral focal ischemia.86 Following SCI, increasing evidence has suggested that the overexpression of NKCC1 and/or downregulation of KCC2 have a significant function in the inflammatory response in the development of chronic NP.87 KCC2 downregulation has also been implicated in functional deficits following SCI. BDNF administration reversed these deficits.88 It has also been demonstrated recently that NKCC1 activity is up-regulated approximately two weeks following cSCI which precedes onset of chronic NP. Immunoblotting showed a significant transient up-regulation of NKCC1 protein in the lesion epicenter of the rat spinal cord on day 14 post-SCI. This was accompanied by a concurrent down-regulation of KCC2 protein. Moreover, inhibition of NKCC1 with its potent antagonist bumetanide (BUM) significantly reduced pain behavior in rats.89
Edemic Response
Edema, along with the inflammation and the immune response, is an important contributor to secondary injury following both brain and SCI.90 Edema can be separated into vasogenic and cytogenic edema. Vasogenic edema is due to disruption of the BSCB integrity and cytogenic edema is due to increased intracellular water content. Since astrocytes outnumber neurons 20 to 1, astrocytic cytotoxic edema is believed to be a significant contributor of total cytotoxic edema due to aquaporin-4 expression.91 However, in order for water to have a net flux in a given direction an osmotic gradient must exist.92 Increasing evidence has suggested that NKCC1 may have a role in modulating this osmotic balance. In the brain, this co-transporter has been shown to play an integral part of edema formation following traumatic injury and ischemia.93 Another study has implicated MMP-9 in brain edema following hemorrhage. It demonstrated that hemoglobin-induced oxidative stress, resulting from the rupture of the blood-brain barrier, can trigger MMP-9 in astrocytes and consequently matrix degradation.94 A possible mechanism by which MMPs can increase vasogenic edema is through up-regulation of IL-1β and TNF-α. These cytokines have been directly implicated in the development of vasogenic edema in brain.95 Studies have also demonstrated significant correlation between increased edema and decreased neurobehavioral parameters.96 The extent of edema is closely related to the degree of SCI-induced motor dysfunction.97 However, whether the edemic response following SCI directly induces NP is still unknown.
Immune Response
Growing evidence is implicating a significant role of the immune response in the development of neuropathic pain, particularly through the activity of immune cells and microglia.98 NP has many features of a neuroimmune disorder. Consequently, therapies involving the immunosuppression and the blockade of the reciprocal signaling may produce more effective results than those aimed primarily at modulating the pain symptoms through attenuating neural activity. Several of the factors released by these immune cells, such as cytokines and neutrophic factors, are known to be involved in the development of NP. In addition to contributing to the inflammatory response, some of these signals can modulate the properties of the afferent sensory neurons.
TNF-α
A number of cytokines are produced following SCI.99 TNF-α is a prominent cytokine produced by activated immune cells and microglia. It is initially synthesized as a transmembrane protein that is then cleaved by protease to yield its active form. TNF-α interacts with a diverse family of TNF receptors and thus serves as an important intermediate in the innate immune response. While inflammation is of primary concern, TNF receptors also can induce cell proliferation, differentiation, and apoptosis.100 Following SCI in mice, TNF-α mRNA expression is induced as early as 15 minutes post-injury.101 TNF-α activity has been correlated with NP development through induction of glial and neuronal apoptosis through a NO-mediated mechanism.102 Evidence also exists implicating TNF-α activity in a neuroprotective role following SCI. Specifically, it is thought to protect against excitotoxic amino acids and facilitate in the maintenance of intracellular Ca++.103
IL-1b
Among the proinflammatory cytokines, IL-1β is particularly known to modulate pain sensitivity. Interleukin-1B (IL-1β), along with TNF-α, is commonly involved in the inflammatory response following CNS injury.99 Central administration of IL-1β was shown to produce thermal hyperalgesia and mechanical allodynia.104 Mutant mice with impaired IL-1β signaling exhibit no neuropathic pain, almost no self-mutilating behavior, and little ectopic neuronal activity, suggesting that IL-1β plays a critical role in the NP response.105 Further, rat antibody to IL-1β receptor reduced thermal hyperalgesia and mechanical allodynia in a dose-dependent fashion in mice following nerve injury.106 Bumetanide administration has been shown to reduce the inflammatory response through an attenuation of IL-1β overexpression.89 Additionally, thiazolidinediones (TZDs), potent synthetic agonists of the transcription factor peroxisome proliferator-activated receptor-gamma (PPAR gamma), were shown to induce neuroprotection after cerebral ischemia by blocking inflammation through IL-1β reduction.107 Il-1β is also known to play an important role in the upregulation of MMPs.24
T-lymphocytes
Recent studies have begun to indicate a significant role of T-lymphocytes in the pathology of NP.108 It is known that these immune cells mediate inflammation following injury through the secretion of pro-inflammatory cytokines although it was not clear whether this produced a beneficial or detrimental outcome. Recent studies have confirmed that recruitment of T-cells produce secondary tissue damage and impair functional recovery.109 Additionally, athymic rats, which lack mature T-cells, develop a significantly reduced mechanical allodynia and thermal hyperalgesia following SCI.110 Other studies have confirmed the importance of T-cells in the onset of NP.111 T-cell infiltration and activation in the dorsal horn of the spinal cord following injury contribute to the evolution of neuropathic pain-like hypersensitivity through interferon-gamma upregulation.112 This same study also showed that IFNg signaling is required for full expression of adult neuropathic hypersensitivity.
Neutrophils
Neutrophils are commonly found in the blood stream and are resident in the peripheral nervous system. They are the first leukocytes to arrive at damaged tissue, and release molecules that can affect neural function.113 They are most numerous 1-3 days after SCI, are detectable for up to 10 days post injury, and heavily express MMP-9.114 Myelin basic protein, a major membrane protein component of the central nervous system, is a cleavage substrate for MMP-9, suggesting it has a supporting role in demyelination and permeability characteristics in the blood brain barrier.115 Disruption of the BSCB allows for neutrophil invasion which can contribute additional tissue damage and demyelination. Invading neutrophils will facilitate lesion expansion via necrotic mechanisms and also through the production of reactive oxygen species, proteases, and nitric oxide.27
Microglia
Microglia are the resident immune cells of the CNS and are continually surveying their environment for foreign markers.116 Being part of the innate immune response, they are quickly activated following injury or infection and share many similarities to the primary immune cell of the PNS, the macrophage.117 Microglia are distinguished from macrophages by their relatively high expression of CD45, reduced antigen presenting functions, and less pronounced inflammatory response. All of the known Toll like receptors (TLRs) are expressed by microglia, allowing them to detect bacterial and viral molecular patterns.118 After a nerve lesion, microglial cells form dense clusters around the cell bodies of injured motor neurons in the ventral horn of the spinal cord, similar to the way macrophages surround injured sensory neurons in the PNS.119 Following activation, inflammatory cytokines, such as TNF-α, IL-1β, IL-6, and NO are expressed along with MHC class II costimulatory molecules, allowing them to activate the adaptive immune response.17 As mentioned previously, acute MMP-9 production is also mediated by microglia activation.28 Three signaling pathways mediate the recruitment of resident spinal microglia and circulating monocytes to the dorsal horn near the injury site. These involve the chemokine fractalkine acting on the CX3CR1 receptor, CCL2 signaling through CCR2, and Toll-like receptors.15,118,120 Microglial activation at the spinal cord contributes to mechanical hyperalgesia and spinal neuronal hyperactivity induced by diabetes, by reducing KCC2 expression.121 In SCI, activated microglia contribute to hyper-resopnsiveness in the dorsal horn by releasing molecules that further stimulate microglia production of inflammatory mediators. This positive feedback loop can further contribute to the development of NP.122 Microglial activation is pivotal in the development and maintenance of below-level allodynia after SCI through mechanisms involving both TNF-α and IL-1β and in chronic states, IL-6.123 CB2 receptors are expressed by peripheral immune cells, including macrophages and lymphocytes, and by microglia and astrocytes in the central nervous system.124 Activation of CB2 receptors located on microglial cells could play a crucial role in limiting the spread of the neuroinflammatory process following SCI through reducing their activation and the consequent release of proinflammatory cytokines.125
Conclusion
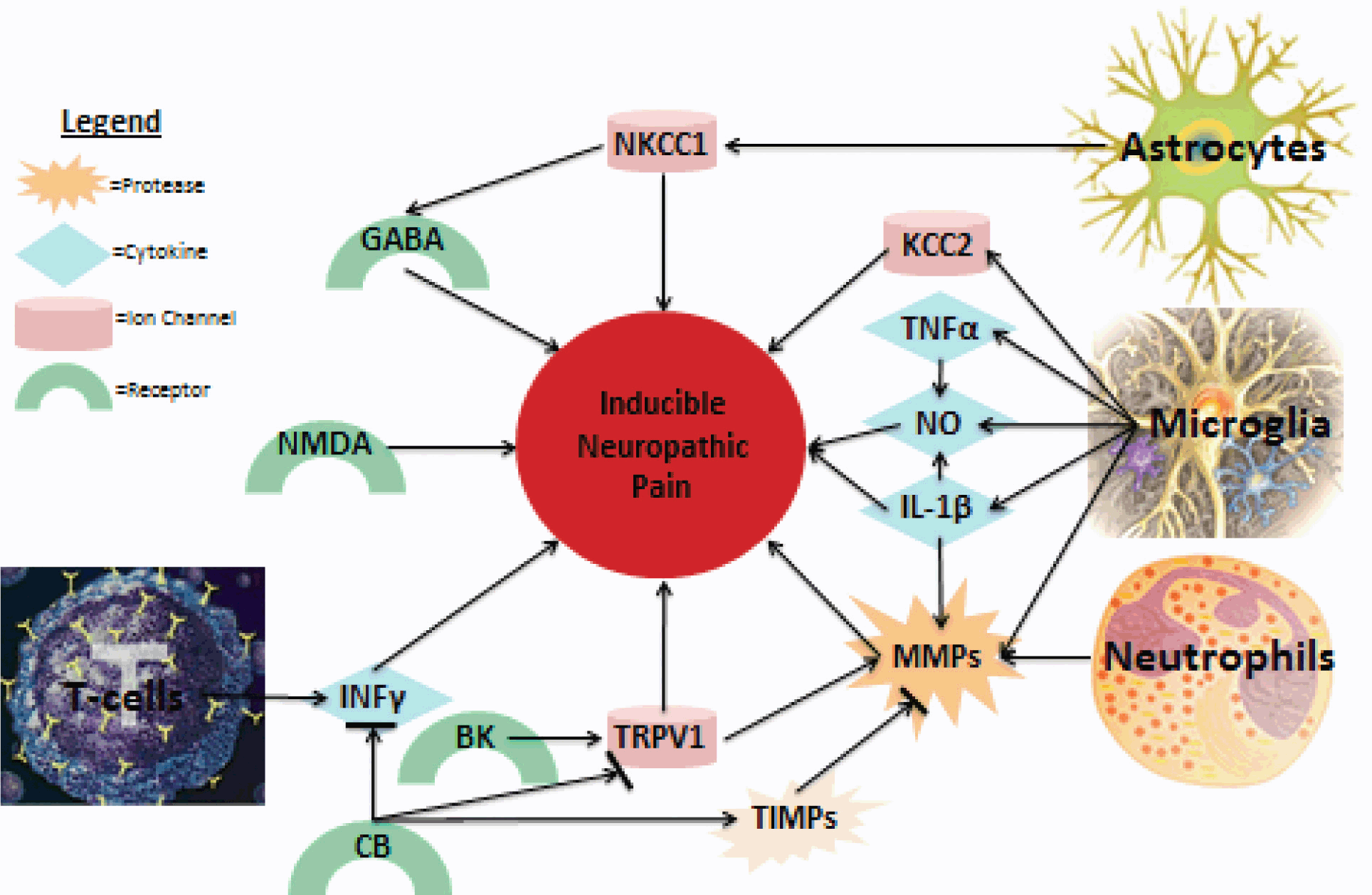
Understanding the spatio temporal pattern of the systems discussed here in relation to the development of neuropathic pain is important in understanding how these systems interact. Figure 1 diagrams a few of the most prominent factors involved in the induction of neuropathic pain. It is clear that several systems are involved in its development; however, studies aimed at understanding how these systems interact are necessary before effective treatments can be produced.
Fig. 1: Summary of major NP contributors.
Figure 1 is a summary of both known and probable links discussed in this review directly contributing to NP. More interactions between the discussed proteins and cells, as well as unmentioned factors, are likely, as interplay between the various contributors to NP following spinal cord injury is complex.
Article complies with International Committee of Medical Journal editor uniform requirements for the manuscripts.
Competing interests – None, Source of Funding – None
Received Date : 7 April 2012
Revised Date : 30 June 2012
Accepted Date : 27 July 2012
References
1. Yezierski RP. Pain following spinal cord injury: the clinical problem and experimental studies. Pain 1996; 68(2–3): 185–194.
2. Vranken JH. Mechanisms and treatment of neuropathic pain. Cent Nerv Syst Agents Med Chem 2009; 9(1): 71–78.
3. Kumru H, Kofler M, Portell E et al. Alterations in excitatory and inhibitory brainstem interneuronal circuits after severe spinal cord injury. J Neurotrauma, 2010; 27(4): 721–728.
4. Seltzer Z, Dubner R, and Shir Y, A novel behavioral model of neuropathic pain disorders produced in rats by partial sciatic nerve injury. Pain, 1990. 43: 205–218.
5. Decosterd I and Woolf CJ. Spared nerve injury: an animal model of persistent peripheral neuropathic pain. Pain 2000; 87(2): 149–158.
6. Tan EC. The oxidative response in the chronic constriction injury model of neuropathic pain. J Surg Res 2009; 152(1): 84–88.
7. Hulsebosch CE, Xu GY, Perez-Polo JR et al. Rodent model of chronic central pain after spinal cord contusion injury and effects of gabapentin. J Neurotrauma 2000; 17(12): 1205–1217.
8. Hemsley KM and Hopwood JJ. Development of motor deficits in a murine model of mucopolysaccharidosis type IIIA (MPS-IIIA). Behav Brain Res, 2005; 158(2): 191–199.
9. Baptista AF, Gomes JR, Oliveira JT et al. A new approach to assess function after sciatic nerve lesion in the mouse - adaptation of the sciatic static index. J Neurosci Methods, 2007. 161(2): p. 259–64.
10. Varejao AS, Meek MF, Ferreira AJ et al. Functional evaluation of peripheral nerve regeneration in the rat: walking track analysis. J Neurosci Methods, 2001. 108(1): p. 1–9.
11. Koka R and Hadlock TA. Quantification of functional recovery following rat sciatic nerve transection. Exp Neurol 2001; 168(1): p. 192–195.
12. Siddall P, Xu CL, and Cousins M Allodynia following traumatic spinal cord injury in the rat. Neuroreport, 1995; 6(9): 1241–1244.
13. Bolcskei K, Petho G, and Szolcsanyi J, Noxious heat threshold measured with slowly increasing temperatures: novel rat thermal hyperalgesia models. Methods Mol Biol, 2010; 617: 57–66.
14. Pezet S and McMahon SB. Neurotrophins: mediators and modulators of pain. Annu Rev Neurosci 2006; 29: 507–538.
15. White FA, Bhangoo SK, and Miller RJ. Chemokines: integrators of pain and inflammation. Nat Rev Drug Discov 2005; 4(10): 834–844.
16. Couture R, Harrisson M, Vianna RM et al. Kinin receptors in pain and inflammation. Eur J Pharmacol, 2001; 429(1-3): 161–176.
17. Popovich PG, Wei P, and Stokes BT. Cellular inflammatory response after spinal cord injury in Sprague-Dawley and Lewis rats. J Comp Neurol, 1997. 377(3): 443–64.
18. Sharma HS. Pathophysiology of blood-spinal cord barrier in traumatic injury and repair. Curr Pharm Des 2005; 11(11): 1353–1389.
19. Hohlfeld R, Kerschensteiner M, Stadelmann C et al. The neuroprotective effect of inflammation: implications for the therapy of multiple sclerosis. Neurol Sci 2006; 27 Suppl 1: S1–7.
20. Correale J and Villa A. The neuroprotective role of inflammation in nervous system injuries. J Neurol 2004; 251(11): 1304–1316.
21. Amantea D Corasaniti MT, Mercuri NB et al. Brain regional and cellular localization of gelatinase activity in rat that have undergone transient middle cerebral artery occlusion. Neuroscience 2008; 152(1): 8–17.
22. Ringshausen I Dechow T, Schneller F et al. Constitutive activation of the MAPkinase p 38 is critical for MMP-9 production and survival of B-CLL cells on bone marrow stromal cells. Leukemia, 2004; 18(12): 1964–1970.
23. del Zoppo, GJ, Alberts MJ, Adams HP et al. Microglial activation and matrix protease generation during focal cerebral ischemia. Stroke 2007; 38(2 Suppl): 646–651.
24. Choi EM and Lee YS. Luteolin suppresses IL-1beta-induced cytokines and MMPs production via p38 MAPK, JNK, NF-kappaB and AP-1 activation in human synovial sarcoma cell line, SW982. Food Chem Toxicol 2010; 48(10): 2607–2611.
25. Kawasaki Y, Wang X, Park JY et al. Distinct roles of matrix metalloproteases in the early- and late-phase development of neuropathic pain. Nat Med 2008; 14(3): 331–336.
26. Ogier C, Bernard A, Chollet AM et al. Matrix metalloproteinase-2 (MMP-2) regulates astrocyte motility in connection with the actin cytoskeleton and integrins. Glia 2006; 54(4): 272–284.
27. Ding, YH Rafols JA, Ding Y et al. Reduced brain edema and matrix metalloproteinase (MMP) expression by pre-reperfusion infusion into ischemic territory in rat. Neurosci Lett 2004; 372(1-2): 35–39.
28. Rosenberg GA, Cunningham LA, Wallace J et al. Immunohistochemistry of matrix metalloproteinases in reperfusion injury to rat brain: activation of MMP-9 linked to stromelysin-1 and microglia in cell cultures. Brain Res 2001; 893(1–2): 104–112.
29. Chang DI, Hosomi N, Lucero J et al. Activation systems for latent matrix metalloproteinase-2 are upregulated immediately after focal cerebral ischemia. J Cereb Blood Flow Metab 2003; 23(12): 1408–1419.
30. Rosell A, Eloy C Arantxa O A et al. MMP-9-positive neutrophil infiltration is associated to blood-brain barrier breakdown and basal lamina type IV collagen degradation during hemorrhagic transformation after human ischemic stroke. Stroke 2008; 39(4): 1121–1126.
31. Dery MA, et al. Atorvastatin prevents early apoptosis after thoracic spinal cord contusion injury and promotes locomotion recovery. Neurosci Lett 2009; 453(1): 73–76.
32. Gantus MA, Nasciutti LE, Cruz CM et al. Modulation of extracellular matrix components by metalloproteinases and their tissue inhibitors during degeneration and regeneration of rat sural nerve. Brain Res 2006; 1122(1): 36–46.
33. Broverman RL, Nguyen KH, da Silveira A et al. Changes in the expression of extracellular matrix (ECM) and matrix metalloproteinases (MMP) of proliferating rat parotid acinar cells. J Dent Res 1998; 77(7): 1504–1514.
34. Vogelezang MG, Scherer SS, Fawcett JW et al. Regulation of fibronectin alternative splicing during peripheral nerve repair. J Neurosci Res 1999; 56(4): 323–333.
35. Nagase H. and Woessner JF. Matrix metalloproteinases. J Biol Chem 1999; 274(31): 21491–21494.
36. Fujimoto M, Takagi Y, Aoki T et al. Tissue inhibitor of metalloproteinases protect blood-brain barrier disruption in focal cerebral ischemia. J Cereb Blood Flow Metab 2008; 28(10): 1674–1685.
37. Reunanen N, Li SP, Ahonen M et al. Activation of p38 alpha MAPK enhances collagenase-1 (matrix metalloproteinase (MMP)-1) and stromelysin-1 (MMP-3) expression by mRNA stabilization. J Biol Chem 2002; 277(35): 32360–32368.
38. Tong, L, Smyth D, Kerr C etal. Mitogen-activated protein kinases Erk1/2 and p38 are required for maximal regulation of TIMP-1 by oncostatin M in murine fibroblasts. Cell Signal 2004; 16(10): 1123–1132.
39. Ramer R and Hinz B. Inhibition of cancer cell invasion by cannabinoids via increased expression of tissue inhibitor of matrix metalloproteinases-1. J Natl Cancer Inst 2008; 100(1): 59–69.
40. Quartilho A, Mata HP, Ibrahim MM et al. Inhibition of inflammatory hyperalgesia by activation of peripheral CB2 cannabinoid receptors. Anesthesiology 2003; 99(4): 955–960.
41. Ibrahim MM, Alexander M Z, Ganesh A T et al. Activation of CB2 cannabinoid receptors by AM1241 inhibits experimental neuropathic pain: pain inhibition by receptors not present in the CNS. Proc Natl Acad Sci U S A 2003; 100(18): 10529–10533.
42. Zhang M, Martin BR, Adler MW et al., Modulation of the balance between cannabinoid CB(1) and CB(2) receptor activation during cerebral ischemic/reperfusion injury. Neuroscience 2008; 152(3): 753–760.
43. Guindon J and Hohmann AG. Cannabinoid CB2 receptors: a therapeutic target for the treatment of inflammatory and neuropathic pain. Br J Pharmacol 2008; 153(2): 319–334.
44. Dom Bourian MG, Turner NA, Gerovac TA et al. B1 and TRPV-1 receptor genes and their relationship to hyperalgesia following spinal cord injury. Spine (Phila Pa 1976) 2006; 31(24): 2778–2782.
45. Racz I, Nadal X, Alferink J et al., Interferon-gamma is a critical modulator of CB(2) cannabinoid receptor signaling during neuropathic pain. J Neurosci 2008; 28(46): 12136–12145.
46. Pol O, Murtra P, Caracuel L et al. Expression of opioid receptors and c-fos in CB1 knockout mice exposed to neuropathic pain. Neuropharmacology 2006; 50(1): 123–132.
47. Sharma HS, Badgaiyan RD, Alm P et al., Neuroprotective effects of nitric oxide synthase inhibitors in spinal cord injury-induced pathophysiology and motor functions: an experimental study in the rat. Ann N Y Acad Sci 2005; 1053: 422–434.
48. Conti A, Miscusi M, Cardali S et al. Nitric oxide in the injured spinal cord: synthases cross-talk, oxidative stress and inflammation. Brain Res Rev 2007; 54(1): 205–218.
49. Arai S, Harada N, Kubo N et al. Induction of inducible nitric oxide synthase and apoptosis by LPS and TNF-alpha in nasal microvascular endothelial cells. Acta Otolaryngol 2008; 128(1): 78–85.
50. Aldieri E, Atragene D, Bergandi L et al. Artemisinin inhibits inducible nitric oxide synthase and nuclear factor NF-kB activation. FEBS Lett, 2003; 552(2-3): 141–144.
51. Chatzipanteli K, Garcia R, Marcillo AE et al. Temporal and segmental distribution of constitutive and inducible nitric oxide synthases after traumatic spinal cord injury: effect of aminoguanidine treatment. J Neurotrauma 2002; 19(5): 639–651.
52. Yu Y, Matsuyama Y, Nakashima S et al. Effects of MPSS and a potent iNOS inhibitor on traumatic spinal cord injury. Neuroreport 2004; 15(13): 2103–2107.
53. Fairbanks CA, Schreiber KL, Brewer KL et al. Agmatine reverses pain induced by inflammation, neuropathy, and spinal cord injury. Proc Natl Acad Sci USA 2000; 97(19): 10584–10589.
54. Yang JY, Kim HS, and Lee JK. Changes in nitric oxide synthase expression in young and adult rats after spinal cord injury. Spinal Cord 2007; 45(11): 731–738.
55. Wu WP, Ongini E, Impagnatiello F et al. A nitric oxide (NO)-releasing derivative of gabapentin, NCX 8001, alleviates neuropathic pain-like behavior after spinal cord and peripheral nerve injury. Br J Pharmacol, 2004. 141(1): 65–74.
56. Gilroy DW, Tomlinson A, and Willoughby DA, Differential effects of inhibition of isoforms of cyclooxygenase (COX-1, COX-2) in chronic inflammation. Inflamm Res 1998; 47(2): 79–85.
57. Resnick DK, Dixon CE, Marion DW et al. Role of cyclooxygenase 2 in acute spinal cord injury. J Neurotrauma, 1998; 15(12): 1005–1013.
58. Zhao P, Waxman SG, and Hains BC. Extracellular signal-regulated kinase-regulated microglia-neuron signaling by prostaglandin E2 contributes to pain after spinal cord injury. J Neurosci 2007; 27(9): 2357–2368.
59. Ferreira J, Beirith A, Mori MA et al. Reduced nerve injury-induced neuropathic pain in kinin B1 receptor knock-out mice. J Neurosci, 2005. 25(9): 2405–2412.
60. Noda M, Kariura Y, Pannasch U et al. Neuroprotective role of bradykinin because of the attenuation of pro-inflammatory cytokine release from activated microglia. J Neurochem 2007; 101(2): 397–410.
61. Rajpal S, Gerovac TA, Turner NA et al. Antihyperalgesic effects of vanilloid-1 and bradykinin-1 receptor antagonists following spinal cord injury in rats. J Neurosurg Spine, 2007. 6(5): p. 420–4.
62. Gunthorpe M.J, Randall A, Davis JB et al. The diversity in the vanilloid (TRPV) receptor family of ion channels. Trends Pharmacol Sci, 2002; 23(4): 183–191.
63. Hagenacker T Ledwig D and Busselberg D. Feedback mechanisms in the regulation of intracellular calcium ([Ca2+]i) in the peripheral nociceptive system: role of TRPV-1 and pain related receptors. Cell Calcium. 2008; 43(3): 215–227.
64. Pitcher MH, Price TJ, Entrena JM et al. Spinal NKCC1 blockade inhibits TRPV1-dependent referred allodynia. Mol Pain 2007; 3: 17.
65. Gavva NR, Anthony WB, Sekhar S et al. Proton activation does not alter antagonist interaction with the capsaicin-binding pocket of TRPV1. Mol Pharmacol 2005; 68(6): 1524–1533.
66. Knerlich-Lukoschus F, Lucius R, Mehdorn HM et al. Spinal Cord Injuries Induce Changes of CB1 Cannabinoid Receptor and C-C Chemokine Expression in Brain Areas Underlying Circuitry of Chronic Pain Conditions. J Neurotrauma 2011; 28(4): 619–634
67. Dickenson AH and Sullivan AF. Evidence for a role of the NMDA receptor in the frequency dependent potentiation of deep rat dorsal horn nociceptive neurones following C fibre stimulation. Neuropharmacology. 1987; 26(8): 1235–1238.
68. Petrenko AB, Yamakura T, Baba H et al. The role of N-methyl-D-aspartate (NMDA) receptors in pain: a review. Anesth Analg 2003; 97(4): 1108–1116.
69. Aley KO and Levine JD. Different peripheral mechanisms mediate enhanced nociception in metabolic/toxic and traumatic painful peripheral neuropathies in the rat. Neuroscience 2002; 111(2): 389–397.
70. Feldblum S, Arnaud S, Simon M et al. Efficacy of a new neuroprotective agent, gacyclidine, in a model of rat spinal cord injury. J Neurotrauma 2000; 17(11): 1079–1093.
71. Yoshimura M. and Yonehara N. Alteration in sensitivity of ionotropic glutamate receptors and tachykinin receptors in spinal cord contribute to development and maintenance of nerve injury-evoked neuropathic pain. Neurosci Res 2006; 56(1): 21–28.
72. Mathiesen O, Imbimbo BP, Hilsted KL et al. CHF3381, a N-methyl-D-aspartate receptor antagonist and monoamine oxidase-A inhibitor, attenuates secondary hyperalgesia in a human pain model. J Pain 2006; 7(8): 565–574.
73. Christoph T, Schiene K, Englberger W et al. The antiallodynic effect of NMDA antagonists in neuropathic pain outlasts the duration of the in vivo NMDA antagonism. Neuropharmacology 2006; 51(1): 12–17.
74. Hempenstall K, Nurmikko T J, Johnson R W et al. Analgesic therapy in postherpetic neuralgia: a quantitative systematic review. PLoS Med, 2005. 2(7): e164.
75. Kontkanen O, Lakso M, Koponen E et al. Molecular effects of the psychotropic NMDA receptor antagonist MK-801 in the rat entorhinal cortex: increases in AP-1 DNA binding activity and expression of Fos and Jun family members. Ann N Y Acad Sci 2000; 911: 73–82.
76. Gwak YS, Tan HY, Nam TS et al. Activation of spinal GABA receptors attenuates chronic central neuropathic pain after spinal cord injury. J Neurotrauma 2006; 23(7): 1111–1124.
77. Bowery NG Hudson AL, and Price GW. GABAA and GABAB receptor site distribution in the rat central nervous system. Neuroscience. 1987; 20(2): 365–383.
78. Hwang JH and Yaksh TL. The effect of spinal GABA receptor agonists on tactile allodynia in a surgically-induced neuropathic pain model in the rat. Pain, 1997. 70(1): p. 15–22.
79. Patel, S Kesingland A, Froestl W, et al. The effects of GABA(B) agonists and gabapentin on mechanical hyperalgesia in models of neuropathic and inflammatory pain in the rat. Pain, 2001. 90(3): 217–226.
80. Gwak YS and Hulsebosch CE, GABA and central neuropathic pain following spinal cord injury. Neuropharmacology 2011; 60(5): 799–808.
81. Eaton MJ Martinez MA, and Karmally S A single intrathecal injection of GABA permanently reverses neuropathic pain after nerve injury. Brain Res 1999; 835(2): 334–339.
82. Sorkin L.S, Puig S, and Jones DL. Spinal bicuculline produces hypersensitivity of dorsal horn neurons: effects of excitatory amino acid antagonists. Pain, 1998; 77(2): 181–190.
83. Cramer SW Christopher B, John C et al. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol Pain 2008; 4: 36.
84. Hasbargen T, Ahmed MM, Miranpuri G et al. Role of NKCC1 and KCC2 in the development of chronic neuropathic pain following spinal cord injury. Ann N Y Acad Sci 2010.; 1198: 168–172.
85. Ben-Ari, Y. Excitatory actions of gaba during development: the nature of the nurture. Nat Rev Neurosci 2002; 3(9): 728–739.
86. Chen H and Sun D. The role of Na-K-Cl co-transporter in cerebral ischemia. Neurol Res 2005; 27(3): 280–286.
87. Price TJ Hammond DL, Prescott SA et al. Chloride regulation in the pain pathway. Brain Res Rev 2009; 60(1): 149–70.
88. Boulenguez P, Liabeuf S, Bos R et al. Down-regulation of the potassium-chloride cotransporter KCC2 contributes to spasticity after spinal cord injury. Nat Med 2010. 16(3): 302–307.
89. Lu KT, Peng JH, Wang CL et al. Bumetanide administration attenuated traumatic brain injury through IL-1 overexpression. Neurol Res 2007; 29(4): 404–409.
90. Saadoun S and Papadopoulos MC. Aquaporin-4 in brain and spinal cord oedema. Neuroscience 2010; 168(4): 1036–1046.
91. Nesic O, Ye Z, Unabia GC et al. Acute and chronic changes in aquaporin 4 expression after spinal cord injury. Neuroscience 2006; 143(3): 779–792.
92. Kahle KT, Simard JM, Staley KJ et al. Molecular mechanisms of ischemic cerebral edema: role of electroneutral ion transport. Physiology (Bethesda) 2009; 24: 257–265.
93. Jayakumar AR, Panickar KS, Curtis KM et al. Na-K-Cl Cotransporter-1 in the Mechanism of Cell Swelling in Cultured Astrocytes After Fluid Percussion Injury. J Neurochem 2011; 117(3): 437–448.
94. Tejima E, Tsuji K, Rosell A et al. Astrocytic induction of matrix metalloproteinase-9 and edema in brain hemorrhage. J Cereb Blood Flow Metab 2007; 27(3): 460–468.
95. Holmin S and Mathiesen T Intracerebral administration of interleukin-1beta and induction of inflammation, apoptosis, and vasogenic edema. J Neurosurg 2000; 92(1): 108–120.
96. Nout YS, Mihai G, Tovar T et al. Hypertonic saline attenuates cord swelling and edema in experimental spinal cord injury: a study utilizing magnetic resonance imaging. Crit Care Med 2009; 37(7): 2160–2166.
97. Sharma HS, Mohanty S, Wiklund L et al. Post-injury treatment with a new antioxidant compound H-290/51 attenuates spinal cord trauma-induced c-fos expression, motor dysfunction, edema formation, and cell injury in the rat. Acta Neurochir Suppl 2006; 96: 322–328.
98. Scholz J and Woolf CJ The neuropathic pain triad: neurons, immune cells and glia. Nat Neurosci 2007; 10(11): 1361–1368.
99. Pineau I and Lacroix S. Proinflammatory cytokine synthesis in the injured mouse spinal cord: multiphasic expression pattern and identification of the cell types involved. J Comp Neurol 2007; 500(2): 267–285.
100. Hehlgans T and Pfeffer K. The intriguing biology of the tumour necrosis factor/tumour necrosis factor receptor superfamily: players, rules and the games. Immunology 2005; 115(1): 1–20.
101. Brewer KL and Nolan TL. Spinal and supraspinal changes in tumor necrosis factor-alpha expression following excitotoxic spinal cord injury. J Mol Neurosci 2007; 31(1): 13–21.
102. Yune TY, Chang MJ, Kim SJ et al. Increased production of tumor necrosis factor-alpha induces apoptosis after traumatic spinal cord injury in rats. J Neurotrauma 2003; 20(2): 207–219.
103. Cheng B Christakos S and Mattson MP. Tumor necrosis factors protect neurons against metabolic-excitotoxic insults and promote maintenance of calcium homeostasis. Neuron 1994; 12(1): 139–153.
104. Watkins LR, Martin D, Maier SF et al. Characterization of cytokine-induced hyperalgesia. Brain Res 1994; 654(1): 15–26.
105. Wolf G, Tal M, Yirmiya R et al. Genetic impairment of interleukin-1 signaling attenuates neuropathic pain, autotomy, and spontaneous ectopic neuronal activity, following nerve injury in mice. Pain 2006; 120(3): 315–324.
106. Sommer C, Lindenlaub T, Toyka KV et al. Neutralizing antibodies to interleukin 1-receptor reduce pain associated behavior in mice with experimental neuropathy. Neurosci Lett 1999; 270(1): 25–28.
107. Park SW et al. Thiazolidinedione class of peroxisome proliferator-activated receptor gamma agonists prevents neuronal damage, motor dysfunction, myelin loss, neuropathic pain, and inflammation after spinal cord injury in adult rats. J Pharmacol Exp Ther 2007; 320(3): 1002–1012.
108. Moalem G, Xu K, and Yu L. T lymphocytes play a role in neuropathic pain following peripheral nerve injury in rats. Neuroscience 2004; 129(3): 767–777.
109. Jones T B, McGaughy V, Fisher LC et al. Passive or active immunization with myelin basic protein impairs neurological function and exacerbates neuropathology after spinal cord injury in rats. J Neurosci, 2004; 24(15): 3752–3761.
110. Potas JR, Zheng Y, Moussa C, Venn M et al. Augmented locomotor recovery after spinal cord injury in the ATHYMIC nude rat. J Neurotrauma 2006; 23(5): 660–673.
111. Zenonos G and Kim JE. A T cell-orchestrated immune response in the adult dorsal spinal cord as a cause of neuropathic pain-like hypersensitivity after peripheral nerve damage: a door to novel therapies? Neurosurgery 2010; 66(4): N24–5.
112. Costigan M, Andrew M, Alban L, et al. T-cell infiltration and signaling in the adult dorsal spinal cord is a major contributor to neuropathic pain-like hypersensitivity. J Neurosci 2009; 29(46): 14415–14422.
113. Borregaard N Sorensen OE, and Theilgaard-Monch K. Neutrophil granules: a library of innate immunity proteins. Trends Immunol 2007; 28(8): 340–345.
114. Fleming JC, Dekaban GA, Marcillo AE et al. The cellular inflammatory response in human spinal cords after injury. Brain 2006; 129(Pt 12): 3249–3269.
115. Michaluk P and Kaczmarek L. Matrix metalloproteinase-9 in glutamate-dependent adult brain function and dysfunction. Cell Death Differ 2007; 14(7): 1255–1258.
116. Nimmerjahn A Kirchhoff F and Helmchen F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005; 308(5726): 1314–1318.
117. Streit WJ. Microglia and macrophages in the developing CNS. Neurotoxicology 2001 22(5): 619–624.
118. Olson JK and Miller SD. Microglia initiate central nervous system innate and adaptive immune responses through multiple TLRs. J Immunol 2004; 173(6): 3916–3924.
119. Hu P, Keay KA, McLachlan EM et al. Immune cell involvement in dorsal root ganglia and spinal cord after chronic constriction or transection of the rat sciatic nerve. Brain Behav Immun 2007; 21(5): 599–616.
120. Verge GM, Watkins LR, Naeve GS et al. Fractalkine (CX3CL1) and fractalkine receptor (CX3CR1) distribution in spinal cord and dorsal root ganglia under basal and neuropathic pain conditions. Eur J Neurosci, 2004; 20(5): 1150–1160.
121. Morgado C, Gomes FL, Marinho MM et al. Minocycline completely reverses mechanical hyperalgesia in diabetic rats through microglia-induced changes in the expression of the potassium chloride co-transporter 2 (KCC2) at the spinal cord. Diabetes Obes Metab 2011; 13(2): 150–159.
122. Hains BC and Waxman SG. Activated microglia contribute to the maintenance of chronic pain after spinal cord injury. J Neurosci 2006; 26(16): 4308–4317.
123. Detloff MR, Popovich PG, Basso DM et al. Remote activation of microglia and pro-inflammatory cytokines predict the onset and severity of below-level neuropathic pain after spinal cord injury in rats. Exp Neurol, 2008; 212(2): 337–347.
124. Stella N. Cannabinoid signaling in glial cells. Glia 2004; 48(4): 267–277.
125. Racz I. Crucial role of CB(2) cannabinoid receptor in the regulation of central immune responses during neuropathic pain. J Neurosci, 2008; 28(46): 12125–12135.