Annals of Neurosciences, Vol 13, No 1 (2006)
Annals of Neurosciences, Volume 13, Issue 1 (January), 2006
EARLY ONSET FAMILIAL ALZHEIMER'S DISEASE: REVIEW OF A PEDIGREE
Address for correspondence:
A. K. Sharma
“SantAshish”, 36,Ram GaliNo.7, Raja Park, Jaipur-302004,
Phone-0141–5121531,2618631
Abstract
Early onset familial Alzheimer's disease is a rare hereditary disorder which is scarcely reported from India. We are reporting a family in which thirteen members are affected between 46 and 56 years of age. Diagnosis of this entity is important to provide correct prognosis and adequate care to patient and to recognize at risk family members by genetic testing after proper counseling. Recognition of this disease in pedigree may provide new horizon in evolution of novel therapeutic measures.
Review of Pedigree
Thirteen members of a family (Fig-1) were affected in three successive generations. Illness started between 46 to 53 years (mean 57 years) of age. Disease was inherited in autosomal dominant mode of inheritance. Most of these patients had insidious onset and progressive memory impairment with disturbance of language and other cognitive functions. We were able to examine 5 affected members of family. Out of them 3 patients are under regular follow up. Presently two of them are mute incontinent and bedridden with fixed flexion deformities of limbs. Seizures are also present in one patient. Proband is 46 years old male, graduate, government servant by occupation, had 2 year duration of progressive intellectual decline, chiefly affecting memory, language and visuospatial functions. DSM-IV criteria were used for diagnosis of dementia and NINCDS criteria for probable Alzheimer's disease. MRI brain (Fig-2) shows generalized cortical atrophy withselective involvement of temporoparietal cortex. SPECT study using 99mTc labeled L, L-ethyle cysteinate dimer(99mTC-ECD) revealed hypo perfusion in parietotemporal lobe (Fig-3). Causes of reversible dementia were excluded by relevant investigations. Genetic study for young onset familial Alzheimer's disease could not performed because of financial constraint.
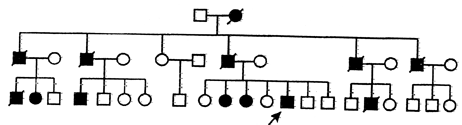
Fig. 1: Pedigree tree of the patient
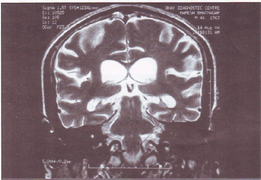
Fig. 2: MM Brain of the patient showing selective atrophy of temporal lobe with generalized cortical atrophy
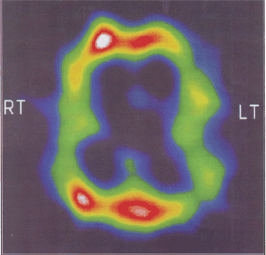
Fig. 3: SPECT of the patient showing hypo perfusion in parietal temporal lobe.
Discussion
Early onset familial Alzheimer's disease is the commonest single cause of young onset dementia followed by vascular dementia and frontotemporal lobar degeneration (4). Familial Alzheimer's disease is a heterogeneous disorder with autosomal dominant mode of inheritance and almost complete penetrance (5).Clinical features of young onset familial Alzheimer's is similar to older onset sporadic Alzheimer's disease. Clinical features tend to follow a characteristic pattern, beginning with memory impairment, spreading to language and visuospatial dysfunctions. Judgment, insight and motivation also declines subsequently. Behavior and social functioning is usually preserved till later stage of disease. Final deterioration leads to bedridden, mute and unresponsiveness, which mimics to persistent vegetative state. Myoclonic jerks and seizures are found occasionally (6). Duration of disease range from 8 to 12 years. Infection and cardiac disease are most common cause of death in such patients. Neuroimaging features can be correlated with clinical features of Alzheimer's disease. Early involvement of hippocampal and entorhinal cortex leads to forgetfulness for daily events. Involvement of parietal lobe manifests as dyspraxia and visuospatial defects. Functional imaging reveals decreased perfusion in posterior temporoparietal cortex. Majority of cases of familial Alzheimer's disease are due to mutation in presenilin-1 gene on chromosome 14. Rarely pathogenic mutations occur in b-amyloid precursor protein gene on chromosome 21 or in presenilin-2 gene on chromosome 1(7). In this pedigree proband also showed similar clinical pattern of involvement. Other causes of dementia was ruled out by relevant investigations. Selective atrophy of temporoparietal lobe in MRI brain along with decrease perfusion on SPECT in parietotemporal lobe points towards the diagnosis of Alzheimer's disease.
Recognition of young onset familial Alzheimer's disease is important to predict natural history of disease and to improve overall care of patient. Despite progress made in understanding the pathogenesis, the development of curative treatment in Alzheimer's disease remains difficult. However identification of genetic aspect and molecular mechanisms in cases of early onset familial Alzheimer's disease is important for determination of prognosis and future production of disease modifying agents.
References
1. Peter Hereda, R Scott Turner. Inherited dementias. Neurol Clin N Am 2002; 20: 779–808.
2. Sanpson E L , Warren J D, Rossor M N. Young onset dementia. Postgrad Med J 2002; 80: 125–139.
3. Kennedy A M, Newman S K, et al. Chromosome 14 linked familial Alzheimer's disease: A clinicopathologic study of A single pedigree.Brainl995; 118: 185–205.
4. Harvey RJ, Rossor MN, Skelton- Robinson M, et al. Young onset dementia: epidimeology, clinical symptoms, family burden support and outcome. London: Dementia Research group, 1998.
5. Zekanowskic, Religa D, Graff C, Filipek S. Genetic aspects of Alzheimer's disease. Acta Neurobiol Exp(wars) 2004; 64: 19–31.
6. Hauser WA, Morris ML, Heston LL, et al. Seizures and myoclonus in patients with Alzheimer's disease. Neurology. 1986; 36: 1226–1230.
7. Popescu BO, Ankarcrona M. Mechanisms of cell death in Alzheimer's disease: role of presenillins. J Alzheimer's Dis 2004; 6: 123–128.