Annals of Neurosciences, Vol 13, No 1 (2006)
Annals of Neurosciences, Volume 13, Issue 1 (January), 2006
HOMOCYSTEINE: A POSSIBLE MODIFIABLE RISK FACTOR IN VASCULAR DEMENTIA
Corresponding author
Sunil Pradhan
Department of Neurology, Sanjay Gandhi Postgraduate Institute of Medical Sciences, Lucknow
Introduction
Vascular dementia (VaD) is a degenerative cerebrovascular disorder that results from ischemic or hemorrhagic brain damage. Epidemiological studies suggest that in eastern population like China, Japan, and Russia, VaD is more prevalent than AD(1). The five years survival rate is 39% for patients with VaD with respect to 75% for age matched controls. In some studies it has been found that homocysteine levels increase with age. Multi infarct dementia (MID) is the most common type of VaD that leads to a progressive decline in memory and cognition by narrowing blood vessels in the brain. This narrowing is done by accumulation of atherosclerotic plaques in these vessels. Probably this co-existence of VaD with atherosclerotic disease is the cause of higher mortality rate than AD. Hyperhomocysteinemia which is a condition of moderately high plasma level of homocysteine has been shown to be associated with cerebrovascular disease. Hyperhomocysteinemia, now a day, is being considered as an independent and modifiable risk factor for cardiovascular as well as cerebrovascular diseases, besides other risk factors like smoking, dyslipidemia, hypertension and obesity.
A number of studies indicate that after a methionine load test, mild hyperhomocysteinemia occurred in 21%, 24% and 32% of patients with coronary artery disease (CAD), cerebrovascular disease (CVD), and peripheral vascular disease respectively(2). Cerebrovascular diseases in turn may cause dementia. Three months after hospitalization for acute ischemic stroke, 25% to 30% of patients meet criterion for dementia (3,4)
Cognition impairment and homocysteine
VaD is a broad term that includes cognition impairment after a single strategically placed infarct or multiple small cortical infarcts that often go unnoticed and cause damage to the cortex of the brain. Later was named as MID (multi-infarct dementia) by Hachinski et al. in 1974. The disorder is associated with atherosclerotic plaques which damage the lining of arteries. MID is not caused directly by the deposition of atherosclerotic plaques but by a series of strokes that leaves areas of dead brain cells (infarcts). In experimental models of atherogenesis it has been shown that increased homocysteine concentration in blood along with dietary lipids produces lesion of the intima.(5) It has also been shown that high levels of homocysteine induce sustained injury of arterial endothelial cells that accelerates the development of thrombosis and atherosclerosis. (6) When laboratory animals were infused with 0.1–0.3 mM homocysteine, their arteries showed a thicker intima, proliferation of their smooth muscle cells, increased desquamation (detachment of cells in the luminal surface of arteries or veins) of the luminal surface and high levels of foam cells.(7) This proliferative effect of homocysteine on arterial smooth muscle cells is a relevant feature of atherosclerosis. (8,9) Homocysteine has also been shown to increase DNA synthesis in vascular smooth muscle cells, while impeding the regeneration of damaged endothelial cells through an inhibitory effect on endothelial cell growth.(10,11) It is also likely that homocysteine interacts with other growth factors and cytokines present in atherosclerotic lesions (12) to promote the growth of smooth muscle cells during atherogenesis. (13)
Homocysteine also facilitates the generation of hydrogen peroxide (H2O2).(14) By creating oxidative damage to LDL cholesterol and endothelial cell membranes, hydrogen peroxide can catalyze injury to vascular endothelium. (14,15) It thus renders Nitric oxide (NO2) more susceptible to oxidative damage. NO2 and other oxides of nitrogen released by endothelial cells protect endothelial cells from damage by reacting with homocysteine, forming S- nitrosohomocysteine, which inhibits hydrogen peroxide formation. However as homocysteine level increases this protective mechanism can be overloaded allowing damage to endothelial cells. (16,17,18) Also sulfate compounds play an important role in the formation of amino sugars required to form the basement membrane of blood vessels. High levels of homocysteine are likely to contribute to the formation of abnormal blood vessels which are more susceptible to damage. End result of these events is the formation of atherosclerotic plaques.
Homocysteine has also been found to be a neurotoxin especially in conditions in which glycine levels are elevated, including head trauma, stroke, and B12 deficiency. (19) Homocysteine interacts with the N-methyl- D- aspartate receptor, causing excessive calcium influx and free radical formation, resulting in neurotoxicity (19). The neurotoxic effect of homocysteine and / or reduced methylation reaction in the CNS contributes to the mental symptomatology seen in B12 and folate deficiency. Significant decrease in B12 and folate are common in the elderly population and can contribute to a decline in cognitive function (20,21,22). An investigation of cognitive ability in older men found poorer spatial copying skills in those individuals with higher homocysteine levels. Better memory performance was correlated with higher vitamin B6 levels. (23) B12 deficiency and increasing severity of cognition impairment has been seen in Alzheimer's disease patients compared to controls and patients with other dementias. (24) In a study of 741 psycho geriatric patients, high plasma homocysteine levels were found in dementia and non demented patients; however only demented patients also had lower blood folate concentrations compared to controls. Patients with concomitant vascular disease had significantly higher plasma homocysteine than those without diagnosed vascular disease.(25)
Normal homocysteine metabolism
Homocysteine is sulfur containing non-proteinogenic amino acid biosynthesized from methionine. It has two main metabolic fates: the remethylation pathway where it is remethylated to methionine, and trans-sulfuration pathway which degrades homocysteine to cysteine and taurine. Methionine is a limiting amino acid in the synthesis of many proteins and its metabolism affects several biochemical pathways involving production of nutrients which are essential to the optimal functioning of cerebrovascular, skeletal and nervous system. Cysteine and taurine are important nutrients for cardiac functions hepatic detoxification functions, cholesterol excretion, bile salt excretion and glutathione production. Thus, if remethylation or transsulfuration pathway is impaired it will affect all methyl and sulfur group metabolisms occurring in the body. (Figure 1)
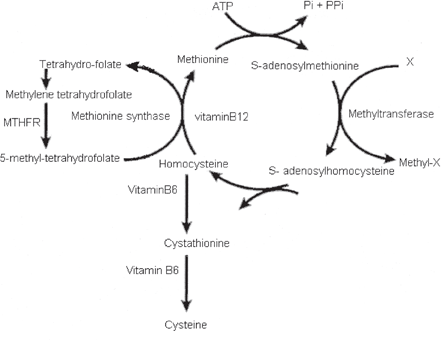
Fig. 1 Homocysteine metabolism. See the text for further details.
The remethylation Pathway:
S-adenosylmethionine (SAM) that is universal methyl donor of the body is synthesized by transfer of an adenosyl group from ATP to the sulfur atom of methionine. SAM transfers its methyl group to an acceptor atom forming S-adenosyl homocysteine (SAH). This SAH on hydrolysis forms adenosine and homocysteine. Homocysteine when remethylated by transfer of a methyl group from 5-methyl tetra hydrofolate catalyzed by methionine synthase, an enzyme that requires vitamin B12 as a cofactor, methionine is formed. Homocysteine level is increased when conversion of homocysteine to cystathione or methionine is impaired due to vitamin B6 or B12 deficiency.
The trans-sulfuration pathway:
Cysteine is biosynthesized from methionine through this pathway. First three steps of the pathway i.e. conversion of methionine to S-adenosyl homocysteine are shared by remethylation pathway. Homocysteine is then acted upon by vitamin B6 dependent enzyme Cystathione β synthase which catalyses its condensation with serine to form cystathione. This is a critical step in the pathway because it is reversible under physiological condition. Cystathione is then cleaved by cystathione synthase, another vitamin B6 dependent enzyme to form oxobutyrate and cysteine. Excess cysteine is oxidized to taurine and inorganic sulfates. Thus in addition to the synthesis of Cysteine this pathway can catabolize effectively potentially toxic excess homocysteine that is not required for methyl transfer. Recently an alternative pathway for conversion of homocysteine into methionine has been proposed.(26) This biochemical pathway involves homocysteine thiolactone formation.(27)
Remethylation and trans-sulfuration pathway are very important for the biosynthesis of some important nutrients which are used to improve risk factor markers and reduce oxidative stress. Dietary antioxidants and folic acid may play a role in the pathophysiology of coronary disease and stroke. Antioxidants (like Ascorbic acid, vitamin E, lycopene, carotene, green tea etc) have been shown to have dramatic effects on preventing strokes, both new and recurrent. (28) There are two main mechanisms that provide primary stroke protection: LDL oxidation prevention and prevention of hyperhomocysteinemia. (29) Three categories of agents which provide this protection against stroke are: 1) dietary antioxidants (vitamins B-complex, C, E, beta-carotene and lycopene), 2) vitamin B12 and 3) the flavanoids. Dietary antioxidants, such as vitamins E, C, B-complex, beta-carotene and lycopene(30,31) have been shown to be important in blocking critical steps in the formation of atherosclerotic plaques. It has already been shown that atherosclerotic plaques are formed from free-radical oxidation of lipids, thereby forming fatty plaques on vessel walls, that leads to vascular stenosis. Vitamin C, a water-soluble vitamin has been important in reducing the free-radical formation in aqueous phase.(32) Beta carotene and vitamin E have been shown to block free radical oxidation in the lipid- phase of LDLs. Vitamin E reduces monocyte adhesion to endothelium, inhibits platelet activation in vitro and inhibits atherosclerosis in rodents. In humans, angiographic and ultrasonographic studies suggest that vitamin E also inhibits the progression of atherosclerosis.(33) The prospective observational evidence is fairly consistent for a protective effect of vitamin E against atherosclerosis.(34) Vitamin B12 is another antioxidant that has recently been identified in the prevention of atherosclerotic plaques is vitamin B12. It is a vitamin that is not found in common vegetable sources, and is often formed in both small and large intestinal bacteria, though it is only absorbed in the small intestine. It is said that those who are vegetarian tend to absorb vitamin B12 at higher rates than do those who are non-vegetarian.(35) Low serum vitamin B12, or genetic defect in uptake or utilization of B12 proteins, such as an intrinsic factor deficiency, can lead to a condition known as hyperhomocysteinemia, which in turn creates a problem in sulfur-amino acid metabolism.(36) Hyperhomocysteinemia increases the risk of stroke as it causes an increased formation of a cyclic reactive form of homocysteine that can react with low-density lipoproteins. Such oxidation can lead to atheroma formation, (macrophagocytic lipid aggregations, secondary to increased LDL uptake by these macrophages), as well as intimal injury, oxidation of cholesterol and unsaturated lipids, platelet aggregation, thrombogenic factors, myointimal hyperplasia, fibrosis, and calcification of atherosclerotic plaques(37). The chronic effects of the buildup of these toxic thiolactone derivatives of sulfur amino acid metabolites can lead to ongoing vascular stenosis, leading to ischemic diseases of many organs, especially cerebral and cardiac tissues. Extra dietary vitamin B can also afford a drop in thrombomodulin, an important factor in the clotting cascade. The drop in thrombomodulin levels reflect decreased vessel endothelial injury, which prevents induction of the clotting cascade.(38)
Studies have shown that supplementation with antioxidant vitamins within 12 h of onset of acute ischemic stroke increased antioxidant capacity, reduced lipid peroxidation products and may have an anti-inflammatory effect. (39–41) Green tea catechins possess potent anti oxidative properties, and the preventive effects against various oxidative diseases have been reported. It has also been shown that daily intake of green tea catechins efficiently protects the penumbra from irreversible damage due to cerebral ischemia, and consequent neurological deficits.(42,43)
Pyridoxal 5 Phosphate (P5P) is the active coenzyme form of vitamin B6 (pyridoxine). This cofactor is involved in myriad biological processes, including the trans-sulfuration pathway of homocysteine. This degradation pathway involves a two-step process resulting in the formation of cystathionine and its subsequent cleavage to cysteine. Both of the enzymes involved, cystathionine synthase and cystthioninase, require P5P as a cofactor. The first step in the degradation of homocysteine, via cystathionine synthase, also requires serine, a downstream metabolite of betaine. Once cysteine is generated it can be directed into several pathways, including synthesis of glutathione, acetyl- CoA, and taurine. There are three known pathways from cysteine to taurine; all require P5P Vitamin B6 administration has also been shown to effectively reduced homocysteine levels and thereby improved the vasodilation. Some studies have also shown that consumption of 1 cup fortified breakfast cereal daily significantly increased B vitamin and decreased homocysteine concentrations, including post-methionine-load homocysteine concentrations. (44, 45) If a dietary deficiency or increased demand resulting from genetic or biochemical alteration exists for methionine, pyridoxine and methylcobolamine, treatment with these micronutrients should reduce homocysteine levels. Several studies utilizing folic acid, B12, B6 and betaine either alone or in combination have demonstrated the ability of these nutrients to normalize homocysteine levels.(46–49)
In a recent placebo- controlled clinical study of hundred men with hyperhomocysteinemia, oral therapy with 650 meg folic acid, 400 meg vitamin B12, 10 mg vitamin B6, or a combination of the three nutrients was given daily for six weeks. Plasma homocysteine was reduced 41.7% (p< 0.001) during folate therapy and 14.8% (p< 0.01) during B12 therapy while 10 mg vitamin B6, did not reduce plasma homocysteine significantly. The combination worked synergistically to reduce homocysteine levels 49.8%32. Oral folate therapy (2.5mg) reduced this hyperhomocysteinemia in 94% of treated patients (mean decrease 27%).(50) Folates function as methyl donor to create methylcobalamin which is used for re-methylation of homocysteine to methionine.
Methylcobalamin's only known biological function in humans is in the re-methylation of homocysteine to methionine via the enzyme methionine synthase. In order to originally form methylcobalamin from cyanocobalamin or other Cob(III)alamin or Cob(II)alamin precursors, SAM must be available to supply a methyl group. Once methylcobalamin is formed, it functions in the regeneration of methionine by transferring its methyl group to homocysteine. Methyl-cobalamin can then be regenerated by 5-methylTHF.
Choline also reduces homocysteine levels, albeit indirectly. When choline is ingested (the average daily intake from food is about 400–900 mg), some of it is converted to a nitrogenous acid called betaine, and betaine regulates homocysteine by converting it back to methionine, in a process called methylation. It has long been in clinical use (intravenous administration) for stroke treatment in Europe and Japan, and has shown a great positive response in the treatment.(51)
Selenium is a nutritional antioxidant, especially because it is required for the activity of selenium-dependent glutathione peroxidase (a crucial enzyme in hydrogen peroxide detoxification).(52)
Symptomatology of Vascular dementia:
Due to a variety of pathogenic mechanisms involved in vascular dementia, patients manifest a wide spectrum of symptoms that have abrupt onset. These symptoms include aphasia, abnormal executive functioning, impaired psychomotor performance, changes in personality and mood, gait disturbances, hyperreflexia, extensor plantar response, urinary incontinence, hemiparesis, visual problems, pseudo bulbar syndromes (dysarthria, dysphagia, emotional lability, etc).(53)
Molecular biology of vascular dementia (VaD)
Very less genetic markers are studied in context with vascular dementia, but, clear association between them is still lacking. Some of the important genetic markers which have been shown to be associated with vascular dementia in most of the world literature, are quickly reviewed here.
MTHFR gene – (chromosome 1p36.3)
This gene codes for Methylenetetrahydrofolate Reductase. This enzyme plays an important role in metabolism of homocysteine. Specifically MTHFR converts 5,10-Methylene Tetrahydrofolate to 5-Methyl Tetrahydrofolate, which is required for conversion of homocysteine to methionine. A mutation in this gene produces a defective thermo labile enzyme whose activity is reduced at higher temperature, resulting in increased level of homocysteine in blood of affected people. Mild to moderate elevation in plasma homocysteine level may occur in subjects homozygous for the thermolabile variant of the enzyme MTHFR (10–13% of the White population) particularly in the presence of suboptimal vitamin status.(54) Approximately 24 mutations in MTHFR gene have been identified in people suffering from homocysteinuria, leading to production of an inactivated enzyme. A specific mutation that has been found common in many populations is replacement of C with T at 677 positions. This variant is commonly called as C677T and is found to increase risk of cardiovascular disease including coronary heart disease and stroke in adults. However, genetic polymorphism in MTHFR with elevated homocysteine level is not a universal finding as individuals who are homozygous for the mutated allele and who have folic acid level above the median, are found to have normal plasma homocysteine level.(55) Homocysteine can also be converted to S Adenosyl Homocysteine, which acts as an inhibitor of monoamine neurotransmitter metabolism. (56) From this it may be concluded that raised homocysteine level have the potential to affect neurodegeneration. Association of this gene with plasma homocysteine level and their association with stroke, vascular dementia and AD is debatable (57–60) and need to be confirmed in the context of Indian population.
DCP-1 gene – (Chromosome 17q23)
This gene codes for Angiotensin converting enzyme (a dicarboxypeptidase), which is a membrane, bound ectoenzyme. It converts Angiotensin 1 to Angiotensin 2. Later controls fluid and electrolyte balance, arid systemic blood pressure. Deletion or insertion polymorphism of a 250 bp DNA fragment in intron 16 of DCP 1 gene has been reported.(61, 62) Some studies also support association of ACE activity and amyloid peptide metabolism.(63) ACE deletion allele is also reported to be strongly and independently associated with coronary atherosclerosis & myocardial infarction.(64) but an increase in patients with AD.(65) There are mixed reports for association of ACE gene with Vascular Dementia (66–70), which needs to be confirmed in case of Indian population.
Apo E gene – (Chromosome 19ql3)
This gene encodes apolipoprotein. It provides structural stability to lipoproteins and determines the metabolic fate of particles upon which they reside. In humans three common polymorphisms have been described:
- e3: This gene is present in approximately 75% of Caucasians. Here Cysteine is present at codon 112.
- e4: This form is present in approximately 15% of Caucasians. Arginine is substituted for Cysteine at codon 112.
- e2: This form is present in approximately 10% of Caucasians. Cysteine is present at codons 112 and 158.
Single amino acid differences among common protein isoforms result in significant differences in biochemistry and cellular metabolism of these proteins and marked differences in the risk of AD. Apo E4 allele is over represented in sporadic and familial AD and is a significant risk factor for the disease while Apo E2 is underrepresented and thus may have a protective effect. Studies show consistent increase in frequency of E4 allele in patients with AD and a small reduction in frequency of E2 allele (71). It has also been observed that genotype at APO E accounts for some of the variation in age of onset in population carrying β APP Val 717 Ile mutation.(72,73) Apo e4 gene & head trauma are also found to be a combined risk factors for AD.(74) It has been reported that e4 allele is associated with inadequate neuronal repair and deposition of β amyloid after injury.(75)
ApoE promoter polymorphism A to T and G to T at positions 491 and 219 upstream of the promoter have been found to be a risk factor for AD in many population specially Finnish (Finland).(76) It has also been found that amount of Aβ deposition in APP V 717 F transgenic mouse brain correlates with the level of Apo E expression.(77) Another polymorphism in promoter region of Apo E was found 186 bp upstream of TATA box of Apo E (G to T).(78) This position is a potential binding site for Transcription Factor TH1/ E47.(79) Mutation at this site may affect the regulation of Apo E expression. ApoE4 allele has also been found to be associated with multi infarct dementia. Shimono(80) et al demonstrated an increased frequency of the Apo ≡ 4 allele in patients with MID (20.8%) with respect to that with Japanese control (8.6–11.7%). Noguchi et al reported that frequencies of Apo ≡ 4 both in patients with AD (27.6%) and in patients with MID (21.2%) were significantly higher than the frequency in controls (9.3%) and no significant difference was found between AD and MID. Since Apo≡4 allele is responsible for lipid transport, it is a genuine reason that it may play a role in atherosclerosis.
Presenilin -1 gene- (Chromosome 14)(81,82)
This gene is highly conserved in evolution, being present in C.elegans(83) and D melanogaster.(84) Normally PS-1 gene encodes a polytopic integral membrane protein S-182. The gene is transcribed at low levels in many different cell types both within the CNS and non-neurological tissues. Studies of PS -1 protein in brain and many other peripheral tissues reveal that only very small amount of PS -1 haploprotein exists within the cell at any given time. Despite being present in very small amount, it is actively catabolize by different mechanisms. Normal functions is not clearly defined but it has been speculated that the PS -1 protein might be involved in protein and membrane trafficking, signal transduction and apoptosis. More than 40 different types of mutations have been discovered in PS 1 gene and majority of these mutations are missense mutations giving rise to substitution of a single amino acid. According to a study, it has been found that expression of PS -1 with a M146V mutation in transgenic mice results in altered calcium signaling which may lead to glutamate toxicity to neurons.
Conclusion
Persons with vascular dementia are at greater risk for morbidity and mortality than those with Alzheimer disease.(85) Median survival time after the onset of vascular dementia is 3.3 years, which is much shorter than the 5- 9 years previously estimated.(86) Homocysteine is a classic risk factor for atherosclerosis is expected to have a role in VaD. It interacts with various reactive species and initiates formation of atherosclerotic plaques, thereby increasing the chances of stroke. However, this is one of the modifiable risk factors, as shown by various studies. Increased homocysteine has been shown to have various adverse effects on vascular system both directly and indirectly, either by causing neurotoxicity in specific condition like elevated glycine level, head trauma and stroke or by initiating a cascade of events that lead to atherosclerosis. In both the cases, silent or apparent brain infarctions may occur that lead to cognition impairment that worsens with time, leaving the patient confused, aphasic, emotionally labile, incontinent, and totally dependent on caregivers. Symptomatic treatment for associated psychiatric illness like anxiety, depression, pseudo bulbar palsy effect (inappropriate laughing and crying) should also be done. To prevent the subsequent vascular events, proper management of risk factors is necessary that may include dietary intake of antioxidants, or treatment with micronutrients like methionine, pyridoxine, methylcobalamin etc.
References
1. Fratiglioni L, De Ronchi D, Aguero-Torres H. Worldwide prevalence and incidence of dementia. Drugs Aging 1999; 15(5): 365–75.
2. Boers G. Hyperhomocysteinemia: a newly recognized risk factor for vascular disease. Neth J Med 1994; 45: 34–41.
3. Pohjasvaara T, Erkin Juntii T, Vataja R et al: dementia three months after stroke: baseline frequency and different definitions of dementia in the Helskinki stroke aging memory (SAM) study cohort. Stroke 1997; 28: 785–92.
4. Tatemichi TK, Desmond DW, Stern Y et al. Prevalence of dementia after stroke depends upon diagnostic criterion. Neurology 1992; 42:413.
5. McCully KS, Olszewski AJ, Vezeridis MP. Homocysteine and lipid metabolism in atherogenesis: effect of homocysteine thiolactonyl derivatives, thioretinaco and thioretinamide. Atherosclerosis 1990; 83: 197–206.
6. Harker LA, Slichter SJ, Scott CR, Ross R. Homocysteinemia: vascular injury and arterial thrombosis. N Engl J Med 1974; 291: 537–543.
7. Harker LA, Ross R, Slichter SJ, Scott CR. Homocysteine-induced arteriosclerosis: the role of endothelial cell injury and platelet response in its genesis. J Clin Invest. 1976; 58: 731–741.
8. Ross R. The pathogenesis of atherosclerosis: a perspective for the 1990s. Nature 1993; 362: 801–809.
9. Schwartz SM, Heimark RL, Majesky MW. Developmental mechanisms underlying pathology of arteries. Physiol Rev 1990; 70: 1177–1209.
10. Tsai J, Perrella MA, Yoshizumi M, Hsich CM, Haber E, Schlegel R, Lee M. Promotion of vascular smooth muscle cell growth by homocysteine: a link to atherosclerosis. Proc Natl Acad Sci 1994; USA: 91, 6369–6373.
11. Wang J, Dudman NPB, Wilcken DEL, Lynch JF. Homocysteine catabolism: levels of 3 enzymes in cultured human vascular endothelium and their relevance to vascular disease. Atherosclerosis 1992; 97: 97–106.
12. Stamler JS, Osborne JA, Jaraki O, Rabbani LE, MuUins M, Singel D, Loscalzo J. Adverse vascular effects of homocysteine are modulated by endothelium-derived relaxing factor and related oxides of nitrogen. J Clin Invest 1993; 91: 308–318.
13. Lusis AJ. Atherosclerosis. Nature 2000; 407: 233–241.
14. Stamler J, Slivka A. Biological chemistry of thiols in the vasculature related disease. Nutr Rev 1996; 54: 1–30.
15. Pohjasvaara T, Erkin Juntii T, Vataja R et al: dementia three months after stroke: baseline frequency and different definitions of dementia in the Helskinki stroke aging memory (SAM) study cohort. Stroke 1997; 28: 785–92.
16. Starkebaum G, Harlaun JM. Endothelial cell injury due to copper catalysed hydrogen peroxide generation from homocysteine. J Clin Invest 1986; 77: 1370–6.
17. Stamler J, Osborne J, Jaraki O et al. Adverse vascular effects of homocysteine are modulated by endothelium derived relaxing factors and related oxides of nitrogen. J Clin Invest 1993; 91: 308–18.
18. Stamler J, Loscalzo J. Endothelium derived relaxing factor modulates atherothrombogenic effects of homocysteine. J Cardiovasc Pharmacol 1992; 12: 5202–4.
19. Lipton SA, Kim WK, Choi YB, et al. Neurotoxicity associated with dual actions of homocysteine at the N-methyl-D-aspartate receptor. Proc Natl Acad Sci 1997; 94: 5923–5928.
20. Metz J, Bell AH, Flicker L, et al. The significance of subnormal serum vitamin B12 concentration in older people: a case control study. J Am Geriatr Soc 1996; 44: 1355–1361.
21. Quinn K, Basu TK. Folate and vitamin B12 status of the elderly. Eur J Clin Nutr 1996; 50: 340–342.
22. Fine EJ, Soria ED. Myths about vitamin B12 deficiency. South Med J 1991; 84: 1475–1481.
23. Riggs KM, Spiro A 3rd, Tucker K, Rush D. Relations of vitamin B-12, vitamin B-6, folate, and homocysteine to cognitive performance in the Normative Aging Study. Am J Clin Nutr 1996; 63: 306–314.
24. Levitt AJ, Karlinsky H. Folate, vitamin B12 and cognitive impairment in patients with Alzheimer's disease. Acta Psychiatr Scand 1992; 86: 301–305.
25. Allain P, Le Bouil A, Cordillet E, et al. Sulfate and cysteine levels in the plasma of patients with Parkinson's disease. Neurotoxicology 1995; 16:527–529.
26. Antonio CM, Nunes MC, Refsum H and Abraham AK. A novel pathway for the conversion of homocysteine into methionine in eukaryotes. Biochem J 1997; 328:165–70.
27. Jakubowski H, Protein homocysteinylation: possible mechanism underlying pathological consequences of elevated homocysteine levels. FASEB J; 13: 2277–8.
28. Wilcken D E, Dudman NP Tyrrell PA. Homocystinuria due to cystathine B synthase deficiency – the effects of betaine treatment in pyridoxine responsive patients. Metabolism 1985; 12: 1115–21.
29. Van den Berg M, Boers G, Franken D et al. Hyperhomocysteinemia and endothelial dysfunction in young patients with peripheral areterial occlusive disease. Proc Natl Acad Sci 1997; 94: 5923–5928.
30. VDbergM, BG, Franken D et al. Hyperhomocysteinemia and endothelial dysfunction in young patients with peripheral arterial occlusive disease. Eur J Clin Invest 1995; 25: 176–81.
31. Franken D, Boers G, Blom H et al. Treatment of mild hyperhomocysteinemia in vascular disease patients. Arterioscler Thromb Vase Biol 1994; 14: 465 – 70.
32. Ubbink J, Vermaak W, Van der Merwe et al Vitamin requirements for the treatment of hyperhomocysteinemia in humans. J Nutr1994; 124: 1927 – 33.
33. Landgren F; Israelsson B, Lindgren A et al. Plasma homocysteine in acute myocardial infarction: homocysteine lowering effect of folic acid. J Int Med 1995; 237: 381–8.
34. Diaz MN, Frei B, Vita JA, Keaney JF Jr. Antioxidants and atherosclerotic heart disease. N Engl J Med 1997; 337:408–16.
35. Kevin A Pearce, Maria G. Boosals and Bryan Yeager. Update on Vitamin Supplements for the Prevention of Coronary Disease and Stroke. Am Fam Physician 2000; 62: 1359–66.
36. McCully KS. Chemical Pathology of homocysteine. I. Atherogenesis. Annals of Clinical and Laboratory Science. 1993: 23(6):477–493.
37. Pandey DK, Shekelle R, Selwyn BJ, Tangney C, Stamler J. Dietary vitamin C and betacarotene and risk of death in middle-aged men – The Western Electric Study. American Journal of Epidemiology Dec 1995: 142(12):1269–1278.
38. Rauma AL, Torronen R, Hanninen O, Mykkanen H. Vitamin B-12 status of long-term adherents of a strict uncooked vegan diet (“living food diet”) is compromised. Journal of Nutrition, 1995 Oct, 125(10):2511–5.
39. Parnetti L, Bottiglieri T, Lowenthal D. Role of homocysteine in age- related vascular and non-vascular diseases. Aging, 1997: 9(4):241–57.
40. Selhub J, D'Angelo A. Hyperhomocysteinemia and thrombosis: acquired conditions. Thrombosis and Haemostasis, 1997 Jul, 78(1):527–31.
41. Ullegaddi R, Powers HJ, Gariballa SE. Antioxidant supplementation enhances antioxidant capacity and mitigates oxidative damage following acute ischaemic stroke. Eur J Clin Nutr.2005 Dec; 59(12): 1367–73.
42. Hak AE, Ma J, Powell CB, Campos H, Gaziano JM, Willett WC, Stampfer MJ. Prospective study of plasma carotenoids and tocopherols in relation to risk of ischemic stroke Stroke. 2004 Jul; 35(7): 1584–8.
43. Zhao B. Natural antioxidants for neurodegenerative diseases. Mol Neurobiol 2005; 31(1–3): 283–93.
44. Suzuki M, Tabuchi M, Ikeda M, Umegaki K, Tornita T. Protective effects of green tea catechins on cerebral ischemic damage. Med Sci Monit 2004 Jun; 10(6): BR166–74.
45. Hong JT, Ryu SR, Kim HJ, Lee JK, Lee SH, Yun YP, Lee BM et al. Protective effect of green tea extract on ischemia/reperfusion-induced brain injury in Mongolian gerb ils. Brain Res 2001 Jan 5; 888(1 ):11–18.
46. Kelly PJ, Kistler JR Shih VE , Mandell R, Atassi N, Barron M, Lee H, et al Inflammation, Homocysteine, and Vitamin B6 Status After Ischemic Stroke. Stroke. 2004; 35: 12.
47. Tucker KL, Olson B, Bakun P, Dallai GE, Selhub J, Rosenberg IH Breakfast cereal fortified with folic acid, vitamin B-6, and vitamin B-12 increases vitamin concentrations and reduces homocysteine concentrations: a randomized trial. Am J Clin Nutr 2004 May; 79(5): 805–11.
48. Ansari MA, Ahmad AS, Ahmad M, Salim S, Yousuf S, Ishrat T, Islam F Selenium protects cerebral ischemia in rat brain mitochondria. Biol Trace Elem Res 2004 Oct; 101(l):73–86.
49. Rissanen T, Voutilainen S, Nyyssonen K, Salonen JT. Lycopene, atherosclerosis, and coronary heart disease Exp Biol Med (Maywood).2002 Nov; 227(10): 900–7.
50. Sesso HD, Liu S, Gaziano JM, Buring JE. Dietary lycopene, tomato-based food products and cardiovascular disease in women. J Nutr 2003 Jul; 133(7): 2336–41.
51. Muralikrishna Adibhatla R, Hatcher JF, Tureyen K. CDP-choline liposomes provide significant reduction in infarction over free CDP-choline in stroke. Brain Res. 2005 Oct 5; 1058(1–2): 193–197.
52. Schatz R A, Wilens T E, Sellinger O Z, et al. Decreased transamination of biogenic amines after in vivo elevation of brain S- Adenosyl- L-Homocysteine. J Neurochem 1981; 36: 1739–1748.
53. Chapman Joab, Ningshan Wang, Theresa A et al. ACE MTHFR Factor V Leiden & Apo -E Polymorphism in patients with Vascular and Alzheimer Dementia. Stroke 1998; 29: 1401–1404.
54. Claudio Cortese, Corradino Motti: MTHFR gene polymorphism, homocysteine and cardiovascular disease. Public Health Nutrition; 4 (2b): 493 -497.
55. JA Luchsinger, M X Tang, S Shea et al. Plasma homocysteine level and risk of Alzeimer Disease. Neurology 2004; 62: 1972- 1976.
56. Stephen P, Mc I lroy, Kevin B Dynan et al. Moderately elevated plasma homocysteine, MTHFR genotype & risk of stroke in Northern Ireland. Stroke 2002; 33: 2352–2356.
57. Kehoe PG, Hagit Katzoe Lars Feuk et al. Haplotypes extending across ACE are associated with AD. Human Mol Genet 2003; 12 (8); 859–867.
58. Narain Yolanda, Augustin Yip Terence Murphy et al. Angiotensin Converting Enzyme (ACE) gene and AD susceptibility. J Med Genet 2000; 37: 695–697.
59. Jianguo Hu, Akira Igarashi, Makiko Kamata et al. Angiotensin converting enzyme degrades Alzheimer amyloid β peptide: retards A β aggregation, deposition, fibril formation & inhibits cytotoxicity. The journal of Biochemistry 2001; 276 (51): 47863–68.
60. Eloisa Arbustini, Maurizia Grasso, Roberta Fassini et al. ACE gene deletion allele is independently and strongly associated with coronary atherosclerosis and myocardial infarction. Br Heart J 1995; 74: 584–591.
61. Arregui A, Perry E K, Roser M et al. Angiotensin converting enzyme in Alzheimer Disease: increased activity in caudate nucleus & cortical areas. J Neurochem 1982; 38: 1490–1492.
62. Kehoe PG, Carsten Russ, Stephen Mc il roy et al, Variation in DCP-1 encoding Angiotensin Converting Enzyme (ACE) is associated with susceptibility to AD. Nat Genet 1999; 21: 71–72.
63. Lindsay A Farrer, Tatyana Sherbatich; Sergey A Keryanov et al. Association between Angiotensin Converting Enzyme and AD. Arch Neurol 2000; 57: 210–214.
64. Chapman Joab, Ningshan Wang, Theresa A et al. ACE MTHFR Factor V Leiden & Apo -E Polymorphism in patients with Vascular and Alzheimer Dementia. Stroke 1998; 29: 1401–1404.
65. Saunders A, Strittmatter WJ, Schmechel S, St. George Hyslop P, Pericak Vance M, Joo S H et al Association of Apo e 4 with late onset familial AD and Sporadic AD. Neurology 1993; 43: 1467–72.
66. Sorbi S, Nacmias B, Foreio P and Amaducci L. Epistatic effect of APP 17 mutation and Apo E genotype in Familial AD. Ann Neurol 1995; 38: 124–128.
67. J C Lambert, L Araria Gaumidi, L Myllykangas, C Ellis J C Wang,M J Bullido et al Contribution of Apo E promoter polymorphism to AD risk. Neurology 2002; 59: 59–66.
68. Bales K R, Verina T, Dodel R C et al. Lack of apolipoprotein E dramatically reduces amyloid beta peptide deposition. Nat Genet 1997; 17: 263–264.
69. Jean Charles Lambert, Florennce Pasquier, Dominique Cottel, Bernard Frigard, Philippe Amouyel and Marie- Christine Chartier -Harlin. A new polymorphism in ApoE promoter associated with risk of developing Alzheimer's disease. Hum Mol Genet 1998; 7 (3): 533–540.
70. Hollenberg S M, Sternglanz R, Chang P F and Weintraub H. Identification of a new family of tissue specific basic helix loop helix protein with a two-hybrid system. Mol Cell Biol 1995; 15: 3813–3822.
71. St. George Hyslop P, Haines J, Rogaev E, Mortilla M, Vaula G, Pericak Vance M, et al. Genetic Evidence for a novel Familial AD gene on Chromosome 14. Nat Genet 1992; 2: 330–334.
72. Mayeux R., Ottman R, Maestre G, et al. Synergistic effects of traumatic brain injury & Apolipoprotein e4 in patients with AD. Neurology 1995; 45: 555–557.
73. Sparks DL, Hunsaker JCI, Scheff SW et al. Cortical Senile Plaques in coronary artery disease, ageing & AD. Neurobiology Aging 1990; 11: 601–607.
74. Schellennberg G D, Bird T D, Wizsman E M et al. Genetic linkage evidence for a familial Alzheimer's disease locus on chromosome 14. Science 1992; 258: 668–71.
75. Levitan D, Greenwald I. Facilitation of lin- 12- mediated signaling by Sel 12, a C.elegans S182 AD gene. Nature 1995; 377: 351–354.
76. Bulianne G, Livne – Bar I, Humphreys JM, Rogaev E and St. George Hyslop P. Cloning and Mapping of a close homologue of human presenilin in D meianogaster. Neuroreport 1997; 8(4): 1025–1029.
77. P Frosst, Bloom H J, Milos R, Goyette R Sheppard C A, Matthews R G, Boers G J H et al. A candidate genetic risk factor for vascular disease: a common mutation in methylenetetrahydrofolate reductase. Nat genet 1995; 10: 111–113.
78. Saunders A, Strittmatter WJ, Schmechel S, St. George Hyslop P, Pericak Vance M, Joo S H et al Association of Apo e 4 with late onset familial AD and Sporadic AD. Neurology 1993; 43: 1467–72.
79. Tiret L, Rigat B, Visvikis S, Breda C, Corvol P, Cambien F et al. Evidence, from combined segregation and linkage analysis, that a variant of Angiotensin 1 converting enzyme (ACE) gene controls plasma ACE levels. Am J Hum Genet 1992; 51: 197–205.
80. Wragg M, Hutton M, Talbot C, and the Alzheimer's Disease Collaborative Group. Genetic association between intronic polymorphism in presenilin gene and late onset Alzheimer's disease. The Lancet 1996; 347: 509–512.
81. Sandra E Black. Stroke risk and sequelae define therapeutic approaches. Post Grad Med 2005; 117(1): 165–8.
82. Shimono H, Murase T, Ishibashi S et al. Plasma apolipoproteins in patients with multi infarct dementia. Atherosclerosis. 1989; 79: 257–60.
83. Noguchi S, Murakami K, Yamada N. Apolipoprotein E genotype and Alzheimer disease. Lancet 1993; 342: 737–8.
84. Malinow M R, Nieto F J, Kruger W D, et al. The affects of folic acid supplementation on plasma total homocysteine are modulated by multivitamin use and methylenetetrahydrofolate reductase genotype. Arterioscler Thromb Vase Biol. 1997; 17: 1157–62.
85. Benett D. Public health importance of vascular dementia and Alzheimers disease with cerebrovascular disease. Int J Clin Pract Suppl 2001 ; 120:41–8.
86. Wolfson C, Wolfson D B, Asgharian et al. Clinical progression of dementia study group. A reevaluation of the survival duration after the onset of dementia. N Engl J Med 2001; 344(15): 1111–6.
(c) Annals of Neurosciences.All Rights Reserved