Annals of Neurosciences, Vol 13, No 3 (2006)
Annals of Neurosciences, Volume 13, Issue 3 (July), 2006
THE P53 GENE AND HUMAN VESTIBULAR SCHWANNOMAS
Corresponding Author:
Dr. Rajalakshmi Gope, Ph.D.
Additional Professor
Department of Human Genetics
National Institute of Mental Health and Neurosciences
(NIMHANS) Bangalore 560 029.
Phone: 091 80 2699 5125 Fax: 091 80 2656 4830
Email: rgope@nimhans.kar.nic.in
Abstract
Human vestibular schwannomas (VS) arise from the Schwann cells of the vestibular branch of vestibulo-cochlear nerve or the eighth cranial nerve. Although p53 gene mutations, both germ line and somatic, have been reported in many brain tumors, a scant contribution of p53 gene is speculated for human vestibular schwannomas. However, altered structure and expression of p53 gene and age dependent phosphorylation of p53 protein specifically at the Ser 392 position only in younger patients have been reported in human VS tumors and these results show an important role for p53 in human vestibular schwannomas. A functional NF2 gene and its protein, merlin, are known to be absent in human VS tumors. In the absence of merlin the human VS tumor cells should lack NF2-mediated p53 stabilization. But the p53 protein is shown to be accumulated in both sporadic and familial human VS tumors. A number of pathways have been described that lead to increase in the level of the p53 protein in the VS tumor cells. In this article we have reviewed the structure and function of p53 gene, its possible role in causation of VS tumor and various pathways that leads to increase in the level of p53 protein in the VS tumor cells.
Key words: Tumor suppressor gene, p53 gene, nervous system tumors, human vestibular schwannomas, NF2,
Introduction
Human vestibular schwannomas (VS) arise from the Schwann cells of the vestibular branch of vestibulo-cochlear nerve or the eighth cranial nerve. The annual incidence of these tumors is approximately 1 in 30,000 to 40,000 (1). The p53 gene is one of the most widely studied tumor suppressor (TS) gene. Because of the numerous functions performed by p53 in maintaining the genomic stability and regulating important cellular processes such as the cell-cycle, this gene is described as, “Guardian of the Genome”. The p53 gene has been assigned many cellular functions which can be traced to its transactivation potential that are involved in cell-cycle regulation, apoptosis, development, differentiation, gene amplification, DNA recombination, chromosomal segregation, cellular senescence, nucleotide excision repair and recently in aging (2). In this review we have looked at the p53 gene and its possible functions in human vestibular schwannomas.
Human Vestibular Schwannoma
Vestibular schwannomas (VS) occur in both sporadic and familial forms. The unilateral, sporadic forms account for approximately 90% of the cases and the presence of such a lesion does not generally predispose the patient to develop other neoplasms nor there is any known genetic transmission to the offspring (Figure 1 ). The familial form of vestibular schwannoma is characterized by occurrence of bilateral VS tumors associated with neurofibromatosis type 2 (NF2).
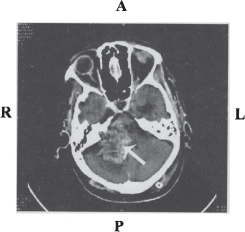
Figure 1 : CT scan image of unilateral vestibular schwannoma
Neurofibromatosis type 2
Robert Smith is credited with first describing a patient with what he called “multiple skin neurogenic tumors”. However, it was Friedrich von Recklinghausen's description in 1882, which made the disease a recognized entity. Von Recklinghausen's disease was initially thought to present in three patterns (3). (a) A predominantly peripheral type (NF1); (b) A predominantly central type in which the occurrence of bilateral vestibular schwannoma was a characteristic feature; and (c) A mixture of peripheral and central types.
Improved understanding led to the formulation of new classification, based primarily on genetic, and somatic differences, at the National Institute for Health Consensus Development Conference in 1987. The proceedings classified the condition as Neurofibromatosis 1 (NF1), previously known as peripheral von Recklinghausen's disease and Neurofibromatosis 2 (NF2), previously known as central von Recklinghausen's disease or bilateral vestibular neurofibromatosis (4).
NF2 occurs in 1 in 30,000 to 40,000 individuals. The hallmark of NF2 is the development of bilateral vestibular schwannomas; however other cranial and spinal tumors may also develop. The mean age at onset of the symptoms is early in the second decade and in most cases it is diagnosed in the late twenties. Males seem to have a milder form of disease than females and the age of onset of symptoms is significantly earlier in maternally affected cases than paternally (1).
Growth rate of vestibular schwannomas
Generally, VS are known to be slow growing tumors. Flow cytometric studies have confirmed a variable mitotic rate in vestibular schwannomas, and that it correlates clinically with the rate of tumor growth (5). DNA cytofluorometric analysis has been used to establish the proportion of cells in S phase of the cell- cycle, but this is not linked to the tumor size or duration of symptoms at the time of presentation. These data confirm nonexistence of any correlation between the tumor size at the time of presentation and the rate of subsequent growth. Hormone receptors like the estrogen and progesterone receptors have been identified in these tumors; however neither of these is fully established to govern the growth behavior of VS. The growth of bilateral vestibular schwannomas associated with NF2 is also variable, but is on average considerably faster when compared to the unilateral VS. NF2s inherited maternally have been known to grow significantly faster compared to paternal inheritance (6).
Age and sex distribution of vestibular schwannoma
Vestibular schwannomas are known to occur predominantly in the middle-ages, however the VS tumors associated with NF2 tend to present earlier with a peak incidence at the third decade (7). It is very rare for vestibular schwannomas to develop in children except for those with NF2 (8).
In adult groups there is a considerable preponderance in women to develop vestibular schwannomas than in men; in children the distribution of these tumors were found to be equal between the sexes. Men seem to have earlier peak prevalence between 36 to 42 years compared to women where peak prevalence is reported to be between 42 to 56 years (7).
Symptoms of VS tumors
Unilateral sensorineural hearing loss, tinnitus and dis-equilibrium are the most common symptoms; other symptoms include mastoid pain or otalgia, headache, facial numbness and diplopia (1,7).
Tumor diagnosis
Magnetic resonance imaging (MRI) is currently the choice for both diagnosis and follow-up of vestibular schwannomas. Intravenous contrast enhancement with gadolinium-DTPA improves the detection rate of small schwannoma which may otherwise have signal intensity similar to brain parenchyma. As only 5% of the patients have normal hearing with good speech discrimination pure tone audiometry forms an important part of evaluation for NF2 (9).
Management of VS
Management of VS is complex and multifaceted because of the high morbidity. In unilateral VS the 7th cranial nerve lies on the periphery of the tumor and can be preserved, whereas in bilateral vestibular schwannomas the 7th and 8th cranial nerves are surrounded by multi lobulated tumor masses between the multiple lobules (10). Very small tumors with useful facial and audio logical function are also closely followed using computed tomography (CT) without surgical intervention. This permits the patient to enjoy a period of hearing as some of these lesions are slow growing and takes many years before surgical intervention becomes essential. Treatment is based on patient's age, occupation, tumor size, current neurological status and growth rate of the tumors.
Non-surgical management of VS
Use of stereotactic cobalt 60 gamma radiotherapy in patients with bilateral vestibular schwannomas has been reported (9). Stereotactic surgery using the “gamma knife” has been advocated by many as a safe and effective alternative to microsurgical removal of these lesions (11). Chemotherapy using doxorubicin hydrochloride, cyclophosphamide and dacarbazine has been described (9,10). Other areas of treatment include rehabilitation, genetic counseling, alternative methods of communication, vocational counseling, screening of first degree relatives, as well as psychiatric guidance.
NF2 gene
The NF2 gene was cloned and mapped to chromosome 22 and it bears sequence similarity to a family of proteins that link the actin cytoskeleton to cell surface glycoproteins, collectively termed as protein 4.1 (12, 13). Among the protein 4.1 molecules the NF2 gene product, merlin or schwanomin is most closely related to Ezrin, Radixin and Moesin (ERM proteins) (14). ERM proteins have been shown to be involved in cellular remodeling. Lack of merlin produces defective early embryonic developmental pathway resulting in failure to initiate gastrulation in mice (15).
Inactivation of NF2 gene and loss of its protein product, merlin, has been reported for all unilateral and bilateral NF2 and related tumors. Merlin is known to function as a negative growth regulator and is shown to bind to bII spectrin and regulate cell morphology and attachment (16).
Recent studies have implicated merlin phosphorylation in regulating merlin sub-cellular localization and growth suppression. P21-activated kinase (PAK) directly phosphorylates merlin at Serine 518. Cyclic AMP-dependent protein kinase is also known to phosphorylate merlin at serine 518 and promotes Merlin-Ezrin hetero-dimerization. This hetero-dimerization is essential in the functioning of merlin, because hypophosphorylation of the merlin protein is known to be crucial for its association with the cell surface trans-membrane proteins such as the CD44. This association is believed to mediate at least one set of signals that are essential in matrix and cell density-dependent growth inhibition (16, 17). Recent studies show merlin as a positive regulator of p53 in terms of stabilization of p53 protein by inhibiting Mdm2 (18).
p53 GENE
p53 protein was first identified during the late 1970s as a cellular protein that formed a tight complex with the SV40 large T antigen and accumulated in the nucleus of cancer cells. The protein was named p53 because it has a molecular weight of 53 kilo Dalton (kDa) (19, 20). Subsequent studies established that the p53 protein formed complexes with other viral proteins such as polyoma virus large T antigens and the adenovirus E1B oncoprotein (21,22). Transfection and expression of p53 gene constructs into cultured normal cells immortalized the cells and transfected p53 gene appeared to cooperate with other oncogenes like H-Ras to immortalize primary embryo fibroblasts. From these studies initially p53 was assumed to be an oncogene and thought to function in a positive fashion to participate in tumorigenesis (23).
One of the evidences to disprove p53 as an oncogene came from the Friend virus-induced mouse erythroleukaemias where p53 gene was found to be a frequent target site for viral integration and many of the integration led to p53 inactivation (22,24). Other evidences include rearrangements and deletions that inactivated p53 completely in HL60 human promyelocytic leukemia line and in several human osteosarcomas (25,26). However, the important finding that disproved p53 to be an oncogene came from further examination of the p53 clones used earlier in the transfection studies. These examinations revealed that the cloned murine p53 used in transfection studies were not of wild type but they carried mis-sense mutation in the coding regions (27). Inactivation of p53 gene in human cancers was demonstrated by the frequent loss of heterozygosity (LOH) of chromosome 17p in a number of tumors particularly colorectal cancers (28). The p53 gene is mutated or lost in approximately 50% of the human tumors studied (29, 30). Germ line mutations occur in individuals with the cancer prone Li-Fraumeni syndrome (31). Recently p53 and its down stream effecter p21 are shown to play key role in the maintenance of genetic stability by regulating error prone DNA repair to yield lower mutation load (32).
Computerized Tomography (CT) scan showing unilateral VS tumor, the arrow indicates the tumor on the right side. Image courtesy Department of Neuroimaging & Interventional Radiology, NIMHANS.
R-right; L-left; A-anterior; P-posterior
Dayalan et al, 2006 (Ref. No. 96); Thomas et al, 2005 (Ref. No. 98)
Structure of p53 gene
The human p53 gene was mapped to chromosome 17p and Southern blot analysis with a variety of probes indicated presence of single copy in the human genome (33). It has 11 exons distributed over 20 kb of DNA and the organization of human p53 gene is similar to the mice p53 gene. The first and second exons are separated by an intron of 10 kb. Restriction enzyme analysis revealed two variant forms of p53 gene having differences in the BglII restriction sites; one containing a 12 kb fragment and the other containing a 12 kb plus a 9 kb fragment and these allelic variants were mapped to intron I region.
p53 gene product
The p53 gene codes for a 3 kb mRNA with an open reading frame of 393 amino acids (33, 34). The p53 protein can be divided into three distinct domains: (i) The amino terminal domain is responsible for strong transactivation function; (ii) The central core region is a DNA binding domain; (iii) The carboxy terminal domain is responsible for tetramerisation of p53 homodimers. The acidic transactivation domain lies within amino acid residues 1–43 (35). The amino terminal domain cooperates with other general transcription factors such as the TATA box binding protein and component of the general transcription factor TFIID during transactivation of target genes (36). The central core of p53 lies within the amino acid residues 100–300 which is responsible for the sequence-specific DNA binding function of the p53 (37,38). p53 protein binds to a consensus-binding site with striking internal symmetry, consisting of 2 copies of a 10 base pair motif separated by 0–13 base pairs and the consensus sequence was identified as 5'-PuPuPu C(A/T)(A/T)G PyPyPy - 3' (39). The carboxyl terminus of the p53 lies within amino acid residues 300–393 that can be further divided into three regions: (a) a flexible linker between residues 300–320 which connects the DNA binding domain to tetramerization domain; (b) tetramerization domain and (c) the extreme carboxyl terminus, which, is a stretch of 30 amino acids, mostly basic. Through X-ray crystallography studies the structure of p53 is shown to be unique consisting of a large beta sandwich that acts as a scaffold for 3 loop-based elements. The sandwich is composed of 2 anti-parallel beta sheets containing 4 and 5 beta strands respectively. The first loop binds to DNA within the major groove, the second loop binds to DNA within the minor groove and the third loop packs against the second loop to stabilize it (40). The most notable feature of the structure is its correlation of data on mutations. The most frequently mutated residues in cancers are all at or near the protein-DNA interface, and over two-thirds of point mutations are in 1 of the 3 DNA loops (41) (Figure 2).

Figure 2: p53 protein structure.
The p53 protein binds DNA as a tetramer and transactivates the expression of down stream genes (42). The tetramerization occurs by interactions between the p53 monomers through the carboxy terminal domain comprising the amino acid residues 325–356 (38). However, when both the mutant and wild type p53 are co-translated, there is a change in the conformation of the wild-type due to the mutant form binding to the wild-type. This change in conformation inactivates the function of wild-type p53 in a dominant negative fashion (43,44).
The 53 kDa nuclear phospho-protein p53 of 393 amino acids comprises of several domains, including an acidic N-terminal region (trans-activation domain), a core region (sequence-specific DNA-binding domain) and a C-terminal domain (with multiple functions).
Adapted from Benchimol et al, 1985 (Ref. No. 33).
Degradation of p53
Under normal conditions the p53 protein is a latent, short-lived protein with a half-life of 15–30 minutes (45). The p53 protein levels and its activity are kept low through various regulatory mechanisms and MDM2 protein is a vital p53 regulatory protein amongst them. MDM2, a 90 kd protein was initially discovered asa gene over-expressed in tumorigenic 3T3 mouse cell line that stably maintains double-minute chromosomes. The amino-terminus of MDM2 binds to the transactivation domain of p53. Although MDM2 association of p53 inhibits the p53-mediated transactivation of target genes (46), the major function of MDM2 is to target p53 for nuclear export (47). The MDM2 functions as an E3 ubiquitin ligase, targeting p53 for degradation through ubiquitin-proteosome degradation pathway (48,49). MDM2 gene itself is a transcriptional target of p53 and is activated in response to UV, suggesting an auto feed-back loop to maintain the p53 concentration within the cell (50).
Activation of the p53 network
The p53 network is normally inactive and is activated only when cells are stressed or damaged. The p53 protein arrests cell division of stressed cells through cell-cycle arrest. In many cases it causes apoptosis of cells in a desperate attempt to contain the damage in order to protect the organism. Early work in this area identified DNA damage as on-switch for p53 activation and recently existence of at least three independent pathways are confirmed through which the p53 network is activated.
(a) The first pathway is triggered by DNA damage due to ionizing radiation. The activation is through two protein kinases ATM and Chk2. ATM is stimulated by double stranded breaks and Chk2 is stimulated by ATM (51).
(b) The second pathway is triggered by aberrant growth signals such as over expression of oncogenes, ras or myc. The p53 network is activated through the pl4 ARF (52).
(c) The third pathway is induced by many factors such as (i) wide range of chemotherapeutic agents (ii) ultraviolet light and (iii) protein kinase inhibitors. ATR and protein kinase II are involved in this pathway (53).
All the three pathways inhibit degradation of p53 protein through various post-translational modifications and stabilize the p53 protein thereby maintaining a high concentration of p53. The stabilised protein undergoes a conformational change, which helps in executing its transactivation function on target genes.
Post-translational modifications and stabilisation of p53 protein
Human p53 protein has been reported to undergo post-translational modifications in at least 18 sites. Seven serines and two threonines in the N-terminal domain of p53, specifically at Ser 6, 9, 15, 20, 33, 37, 46 and threonines 18 and 81 are phosphorylated in response to exposing cells to ionizing radiation or UV light (54). Although post-translational modifications at various sites occur in response to stress, clear differences in responses at individual sites to different agents have been observed. For instance in response to ionizing and UV radiation serine 6,9 and 15 are phosphorylated increasingly as early as 30 minutes after exposure to stress (55). The ATM kinase is the prime kinase for phosphorylating p53 at serine 15 in response to ionizing radiation (56). In response to UV radiation ATR kinase phosphorylates serine 15 and 37 (57). Other kinases that play a role in the phosphorylation at the amino terminus include DNA-PK, which plays an essential role in double strand break repair, phosphorylates p53 at serine 15 (58) and check point kinases Chkl and Chk2 which act down stream of ATM and ATR phosphorylates p53 at serine 20 (59). While ATM and Chk2 act in response to ionizing radiation ATR and Chkl are activated in response to UV irradiation (60). Various chemotherapeutic agents such as actinomycin D also induce serine 15 phosphorylation in a similar manner and stabilize p53 (61). These post-translational modifications stabilise the p53 protein through disrupting the binding of MDM2 and thereby inhibiting the MDM2 induced nuclear export and ubiquitin-mediated proteosomal degradation.
Initial studies focused on amino terminus of p53 and later studies showed importance of other modifications that play a role in p53 stabilization. JNKK kinase, which is expressed in response to UV irradiation, is shown to prolong the half-life of p53 through phosphorylation of p53 at threonine 81 (62). Serine residues in the C terminal regulatory domain are also phosphorylated in response to UV irradiation and these modifications coincide with elevated p53 dependent transcription of target genes. The cyclin B dependent kinase p34 (Cdc2) is involved in phosphorylation of serine 315 in the C terminus of p53 in response to DNA damage by ionizing radiation and chemotherapeutic agents (63,64).
Another important serine residue at position 392 is phosphorylated by PKR in response to interferons released on viral infections (65). p53 is also phosphorylated at Ser 392 by double stranded RNA activated protein kinase and UV light (66). Phosphorylation of p53 at this site coincides with elevated level of p53-dependent transcription (63). Other post-translational modifications such as acetylation at the C terminus are shown to be important for p53 stabilization (67) (Figure 3).
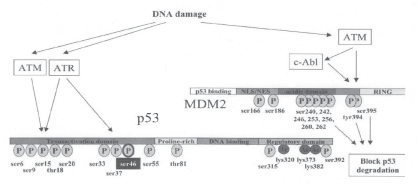
Figure 3: p53 post translational modification
Schematic diagram of the p53 and MDM2 proteins (drawn not to scale). The trans-activation region, proline-rich region, DNA binding domain and the C-terminal regulatory domains of p53 protein are highlighted. Phosphorylation and acetylation sites are indicated by ellipses containing, the letters P and Ac respectively. Detailed description of the various domains are given in the text.
Adapted from Meek et al. 1999 (Ref. No. 53)
Functions of p53 protein
It is well documented that induction of p53 leads to cell growth arrest or cell death. Both provide mechanisms by which p53 functions to control DNA damage, protecting cells from accumulating excessive mutations. Once p53 is stabilized and activated, it induces expression of target genes which helps p53 to carry out its functions. Several genes are controlled directly by p53 and they execute a variety of responses through a network of down stream effectors to bail out the cells from stress.
Role of p53 in cell-cycle inhibition.
The cell-cycle requires coordination of a variety of macromolecular assemblies and movements. Coordination of these complex processes is achieved by cyclin dependent kinases (CDKs). The CDKs form complex with at least two kinds of proteins, the kinase and the cyclin, that drive cells from one stage to the next during cell-cycle which is tightly regulated. p53 plays a major role in cell-cycle arrest in response to DNA damage. At least two check points detect DNA damage: (i) at the G
p53 and apoptosis.
Apoptosis is a biologically important process regulated by a set of genes. The apoptotic cell is characterized by loss of volume, plasma membrane blebbing, nuclear condensation, chromatin aggregation and endonucleolytic degradation of DNA into nucleosomal fragments. These changes are triggered by two major pathways: (i) the death receptor-inducsd extrinsic pathway and the (ii) mitochondria-apoptosome-mediated intrinsic pathway.
Although the ability of p53 to function as a transcriptional activator is necessary for its role in mediating G1 arrest some studies have shown that p53 can induce apoptosis in cells through both transcription-dependent and independent pathways. p53-dependent cell death was shown to occur in presence of either transcriptional or translational inhibitor. In addition, mutant p53 that lacked the transactivational ability also induced apoptosis in cells (69). These data indicate that p53 induces apoptosis in cells independent of its transactivation ability. But other studies where temperature-sensitive mutants of p53 were expressed along with adenovirus E1A protein required the transactivation potential of p53 to induce apoptosis in these cells (70). Thus, it appears that p53 might have two separate pathways to induce apoptosis, one transcription-dependent and the other transcription-independent (71, 72).
Maintenance of genetic stability by p53.
The cellular DNA replication is shown to be error-prone and the genes involved in the repair pathway are known to correct these errors in DNA. When these genes are inactivated the cells accumulate errors in genes leading to genetic instability and predispose cells to tumorigenesis. The p53 protein is important in maintaining genetic stability through transactivation of genes that regulate DNA repair, chromosomal recombination and chromosome segregation and absence of wild type p53 leads to accumulation of DNA errors leading to genomic instability (32,73,74).
Nucleotide excision repair (NER) pathway is an evolutionarily conserved DNA repair pathway with the ability to remove a wide range of DNA adducts induced both by environmental and endogenous sources (75). The NER process is subdivided into two distinct pathways: (i) repair of lesions over the bulk of the genome, referred to as global genomic repair (GGR) and (ii) rapid removal of transcription blocking lesions present in the transcribed DNA strands, known as transcription coupled repair (TCR) (76). Studies using fibroblasts derived from Li-Fraumeni syndrome (LFS), which are homozygous for p53 mutation showed deficiency in GGR following UV irradiation indicating a possible role for p53 in nucleotide excision repair. p53 is shown to regulate nucleotide excision repair by transactivation of NER genes and through direct binding to NER factors. In response to formation of cyclobutane-pyrimidine dimers caused by UV irradiation p53 is shown to bind to NER factors, XPB and XPD, inhibited their helicase activity and thereby regulating nucleotide excision repair (76).
Other studies show the requirement for transactivation function of p53 to regulate nucleotide excision repair. Two specific genes involved in global genomic repair DDB2 and XPC are shown to contain p53 binding consensus sequences (77) and their gene products p48 and XPC respectively were shown to increase in presence of wild type p53 in response to UV irradiation and the global genomic repair of these cells were shown to be enhanced (78).
Although the requirement for p53 in global genomic repair is well established, involvement of p53 in transcription-coupled repair is not well understood. However, one study clearly established the enhancement of transcription-coupled repair in presence of wild type p53, that is, cells with cyclobutane-pyrimidine dimmers induced by UV irradiation (79). Deficiency in base-excision-repair mechanism is also noticed in cells lacking wild type p53 (80) indicating a possible role for p53 in such mechanism of DNA repair as well.
p53 and senescence
Normal mouse or human cells obtained from host undergo limited number of cell divisions in culture followed by senescence. It is shown that p53-deficient murine cells escape senescence and produce immortalized cell lines with aneuploidy. This is most likely to happen due to loss of p53-mediated control over centrosome duplication and the G2-M check point preventing re-initiation of S-phase prior to mitosis or the next G1 phase (81). In addition, it has become clear that p53 responds to signals provided by normal cells undergoing progressive passages in culture. p53 activity increases in late-passage cells, the level of p21 also increases in these cells, slowing or stopping the rate of division of these cultures. Introduction of a transdominant-acting p53 mutant to such cells provides a significant enhancement in the life span of these cells. This clearly indicates a role for wild type p53 in senescence (82).
Inhibition of angiogenesis by p53
To reach enormous sizes tumors induce growth of new blood vessels in their vicinity to bring nutrients required for their aggressive growth. Normal p53 protein stimulates expression of genes that inhibits this process. Therefore, cells with inactivated p53 are more likely to recruit more new blood vessels, providing a critical growth advantage to the tumor cells at a later point during tumor development (82).
p53 in growth and development of the organism
The ability to generate mice lacking p53 implied that p53 is dispensable for growth, differentiation and embryonic development. However, subsequent studies revealed that approximately 16% of 13.5-day p53-/-embryos displayed marked encephaly, with an overgrowth of brain tissue (83). Thus, in contrast to early reports, deficiency of p53 does have a developmental phenotype. Another important finding involves the respective roles of MDM2 and p53 in development. A homozygous deletion of MDM2 results in early embryonic lethality and it is rescued in the absence of p53. These results suggest that the primary role of MDM2 during development is to negatively regulate p53, with p53 and MDM2 acting in concert to regulate cell-cycle during early development (83).
p53 and differentiation
p53 has also been suggested to play a role in differentiation of several cell lineages based on the correlation between over expression of p53 and induction of differentiation markers. p53 is shown to play an important role in the B-cell differentiation and development involving the double strand DNA breaks and rearrangement. The immunoglobulin chains m and k are induced in early pre-B and pre-B cell lines upon expression of p53 and furthermore introduction of mutant p53 was found to block k chain expression in the same cells. Hemoglobin expression is also stimulated in erythroleukemic cells and chronic myelogenous leukemic cells in response to p53 (84). A role in spermatogenesis has also been suggested for p53 based on the highly defined spatial and cyclical expression of the p53 gene in tetraploid pachytene primary spermatocytes.
p53 and tumorigenesis
By the early 1990s p53 was widely recognized as a tumor suppressor gene, as the function of p53 protein was deregulated in most human cancers. In approximately 50% of human tumors p53 gene was found to be inactivated and there are three modes by which mutation of p53 might affect its function: (i) loss of wild type function, (ii) trans-dominant effect of mutant over wild-type p53 function (dominant negative effect) and (iii) gain of oncogenic potential.
The fact that p53 null mice are highly tumor-prone argues strongly that the loss of its function is sufficient to contribute to tumorigenesis. Various factors contribute to loss of wild type function of p53. One possibility is the deletion of one or both p53 alleles, which reduces the expression of tetramers, resulting in decreased expression of the growth inhibitory genes. This mechanism is found in tumors of several types. Another possibility being the non-sense or splice site mutations that result in truncation of the protein that do not allow oligomerization, thus resulting in a similar reduction of p53 tetramers. Mutations of this type are fairly common in cancers of lung, esophagus, and other human cancers. A third mechanism involves mis-sense mutations resulting in dominant-negative effects with an even greater reduction of functionally active tetramers. Such mis-sense mutations are common in cancers of colon, brain, lung, breast, skin, bladder, and other cancers. The dominant-negative effect of mutant p53 proteins through oligomerization with wild type p53 results in an inhibition of the ability of wild type to bind DNA and activate transcription. This is evident from mice carrying dominant-negative trans-gene, which shows increased tumor incidence and decreased survival compared to mice carrying wild-type p53 (85). Thus, presence of mutant p53 proteins in tumors might result from selection for dominant negative mutants that cause loss of wild-type function. A fourth mechanism by which p53 loses its wild-type activity is commonly found in cervical cancers where expression of the E6 gene of human papillomavirus (HPV) results in functional inactivation of p53 through binding and degradation (42).
In addition to inactivation of p53 by mutations in the p53 gene locus some tumors have inactivated p53 by other mechanisms. Increased levels of p53 mRNA and protein have been reported in colorectal carcinomas and many normal and colon cancer cell lines. The p53 over-expression did not depend on the mutational status of the p53 gene (86, 87). In addition, transcriptional modulation of both wild-type and mutant p53 gene has been reported in many normal and tumor tissues in response to DNA damaging agents (88,89). Occasionally tumors with p53 mutants, which predominantly localizes to the cytoplasm have been identified; they lose their ability to act as transcription factors. This novel mechanism of p53 inactivation is identified in breast cancer cells and in a large number of undifferentiated neuroblastomas (90). In approximately one third of all sarcomas the MDM2 gene is amplified, which inactivates the p53 by increasingly marking it for nuclear export and ubiquitin-mediated degradation.
Additionally, some p53 mutants are capable of conferring increased tumorigenicity, metastatic potential and tissue invasiveness. The gain-of-function properties of these mutant p53 proteins may be related to the ability of mutant p53 proteins to preferentially stimulate transcription of several cellular and viral promoters. The mutant p53 proteins may also associate with other cellular proteins like p38, p42 or synergize with PKC in the induction of angiogenic VEGF gene (91).
p53 gene mutations in brain tumors
Patients harboring p53 germ line mutations predominantly develop soft tissue sarcomas and breast cancers, however approximately 13% of them develop brain tumors typically astrocytic glioma. Loss of p53 is well documented in sporadic gliomas glioblastoma multiforme, anaplastic astrocytoma and meningiomas (92).
Germ line mutations are frequently found in patients with multifocal glioma and other malignancies however sporadic p53 mutations are largely documented in astrocytic tumors. There is at least one germline mutation strongly correlated with an unusual accumulation of CNS tumors. The deletion of the codon 236 is associated with a familial brain tumor syndrome (93). This deletion results in a mutant conformation which lack specific DNA binding and transactivation activity which leads to the loss of its tumor suppressor activity.
Despite the presence of many germ line mutations, majority of the mutations of p53 in brain tumors are somatic. The highest number of p53 mutations occurs in regions coding for the DNA binding domain. In majority of human brain tumors the p53 gene is known to undergo a single base pair transition from G:C to A:T(92)
p53 GENE AND HUMAN VS TUMORS
Cancer cells differ from normal cell in many important characteristics, including loss of growth control, differentiation, increased invasiveness and decreased drug sensitivity. These differences arise through a complex process of cellular evolution and it has been suggested that cancer cells have mutations leading to genetic instability and thereby accelerating cellular evolution and studies have identified genes that belong to this category. These genes encode components of cell-cycle checkpoints and ensure an integration of DNA repair and cell-cycle (94). p53 belongs to this category of genes and as described previously it is shown to be mutated in approximately 50% of all human cancers analyzed so far.
Although p53 gene mutations both germ line and somatic, have been reported in many brain tumors, a scant contribution of p53 gene is speculated for human vestibular schwannomas (95). However, altered structure and expression of p53 gene and age dependent phosphorylation of p53 protein specifically at the Ser 392 position only in younger patients have been reported in human VS tumors and these results show an important role for p53 in human vestibular schwannomas (96).
Wild type p53 has a short half-life of approximately 15 minutes and it has auto-regulatory function. However, the p53 protein is stabilized through various post-translational modifications due to DNA damage and stress. The stabilized protein transactivates a set of genes responsible for cell-cycle arrest, DNA repair and apoptosis. Such p53 transactivation leads to elimination of cells carrying damaged DNA through apoptosis pathway. Alternatively, the transactivation helps to correct the DNA damage before committing cells to cell-cycle in order to avoid accumulation of cells with DNA damage.
Mdm2 regulates the level of p53 protein in a cell as it binds p53, ubiquitinates and induces cytosolic-tranlocation where the p53 is degraded through proteosomal pathway. However, NF2 gene product merlin is shown to be a positive regulator of p53 by inhibiting the p53-Mdm2 binding. Thus, in normal cells under stress p53 is stabilized by NF2 in addition to other post-translational modifications (Figure 4a).
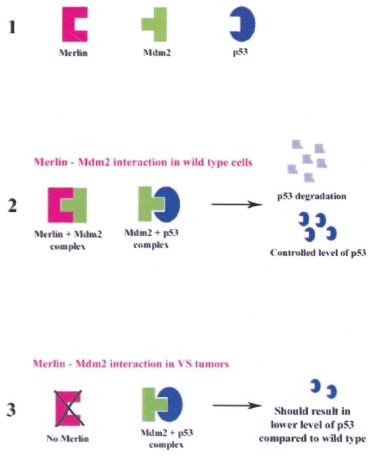
Figure 4a: Merlin Mdm2 and p53 interaction
1 Schematic representation of the proteins merlin, Mdm2 and p53.
2 Stabilisation of p53 by merlin by controling the level of Mdm2.
3 Enhanced degradation of p53 via Mdm2-ubiquitination pathway in the absence of merlin.
Kim et al, 2004 (Ref. No. 18)
However, in many tumor cells p53 protein is known to be accumulated and it could be due to the following mechanism:
(i) Presence of an altered p53 protein in tumors that is unable to bind Mdm2 (Figure 4). Mutant p53 protein acquires altered conformation and it has the ability to form tetramer with wild-type p53 protein, inactivating the later in a dominant negative fashion. Moreover, serine residue at the 392 position could be vital as this is located in the carboxyl terminus among a stretch of 30 basic amino acids. The Ser 392 is known to be phosphorylated under stress due to UV radiation (63) and this could induce a change in the net charge at the C-teminus leading to attaining a possible altered conformation of the p53 protein. Because the p53 monomers tetramerize through the C-terminus, such an altered conformation due to Ser 392 phosphorylation could result in a p53 protein which has altered binding ability to Mdm2, resulting in decreased degradation leading to accumulation of higher levels of p53 protein in tumors.
(ii) Recent reports suggest the existence of wild type p53 and Mdm2 in some lung cancer cell lines, resulting in normal binding between p53 and Mdm2 followed by ubiquitination. But the ubiquitinated p53 does not undergo proteosomal degradation and therefore, p53 protein gets accumulated. It is hypothesized that a possible intermediate required for the degradation of the ubiquitinated-p53 is absent in these cells resulting in the p53 protein accumulation (97) (Figure 4b).
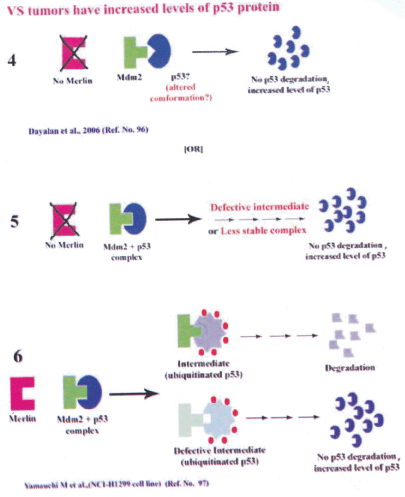
Figure 4b: Possible hypothesis for presence of increased p53 protein level in human VS tumors
4 Mdm2 unable to bind p53 with possible altered conformation resulting in p53 accumulation in the absence of merlin.
5 Defective intemediae of Mdm2 + p53 complex or less stable complex could lead to increased level of p53 protein.
6 Hypothesis put forward by Yamauchi et al 2005 (Ref. No. 97) showing Mdm2-p53 binding and p53 ubiquitination but no p53 degradation due to defective intermediate leading to the accumulation of p53 protein
(iii) In addition, previous report have shown a deregulated pRb pathway in human VS tumors tissues (98). Absence of functional merlin in VS tumors leads to elevated level of CDKs (99) and it results in increased level of hyperphosphorylated pRb (98). The hyperphosphorylated pRb releases free E2Fs in these VS cells, which in turn leads to increased p14ARF. The p14ARF stabilizes the p53 protein by affecting its post-translational modification (phosphorylation) which could lead to decreased degradation resulting in the accumulation of the p53 protein in these human VS tissues. Therefore, a deregulated p53 (96) and pRb (98) pathway could have an important role in the human VS tumor development (Figure 5).
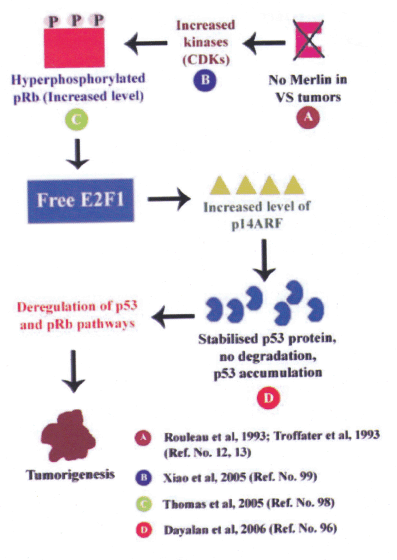
Figure 5: Integration of RB1 and p53 pathways
Absense of merlin in NF2 tumors results in increased CDKs, this leads to increased level of hyperphosphorylated pRb. E2Fs exists as free form in these cells leading to elevated p14ARF and this in turn stabilizes p53 protein. Elevated levels of p53 protein is accumulated in these cells. Thus, deregulated p53 and pRb pathways could lead to human VS tumor development.
(iv) Moreover, results indicate that the percentage of the Ser 392 phosphorylated p53 protein in the VS tumors of the young patients is increased, while the percentage in older patients decreased. Depending on the Ser 392 phosphorylation, the p53 protein could induce alternate pathways in these patients (96) (Figure 6).
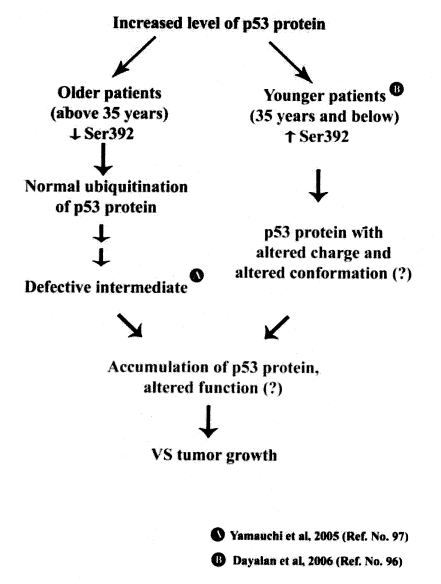
Figure 6: Possible p53 pathways in human VS tumors from older and younger patients
A functional NF2 gene and its protein, merlin, are known to be absent in human VS tumors. In the absence of merlin the human VS tumor cells should lack NF2-mediated p53 stabilization. But the p53 protein is shown to be accumulated in both sporadic and familial human VS tumors (96). Therefore, it could be hypothesized that the increase in the level of the p53 protein in the human VS tumor cells could be due to any of these pathways.
Conclusion
VS tumors are one of the most painful human tumors, one of the most difficult to manage and they cause high morbidity. Extensive studies have been carried out on the possible role of the NF2 gene and its protein, merlin, in these tumors. However, results from recent studies imply that the tumor suppressor genes RBI and p53 could also play important role in the development of these tumors. Further studies are necessary to elucidate the detailed pathways affected by the deregulation of these TS genes. Results from such studies could help us develop novel diagnostic, management and possible treatment procedures for these tumors.
Acknowledgement
We thank all the faculty members of the NIMHANS Neurosurgery department for their help. Part of the results discussed here are from the research project funded by the Department of Biotechnology (DBT), Government of India, to Dr. R.Gope, project number BT/PRO 703/MED/09/133/97. Financial assistance in the form of Junior and Senior Research Fellowships (JRF,SRF) from ICMR (AHPPD), and UGC (MJ and RK) are gratefully acknowledged.
References
1. Martuza RL, Eldridge R. Neurofibromatosis 2 (bilateral acoustic neurofibromatosis). N Engl J Med 1988; 318: 684–88.
2. Oren M, Rotter V. Introduction: p53-the first twenty years. Cell Mol Life Sci 1999; 55: 9–11.
3. Holt GR. Von Recklinghausen's neurofibromatosis. Otolaryngol Clin North Am 1987; 20: 179–93.
4. Seizinger BR, Martuza RL, Gusella JF. Loss of genes on chromosome 22 in tumorigenesis of human acoustic neuroma. Nature 1986; 322: 644–47.
5. Wennerberg J, Mercke U. Growth potential of acoustic neuromas. Am J Otol 1989; 10: 293–96.
6. Markwalder TM, Waelti E, Markwalder RV. Estrogen and progestin receptors in acoustic and spinal neurilemmomas. Clinicopathologic correlations. Surg Neurol 1986; 26: 142–48.
7. Eldridge R. Central neurofibromatosis with bilateral acoustic neuroma. Adv Neurol 1981; 29: 57–65.
8. Allcutt DA, Hoffman HJ, Isla A, et al. Acoustic schwannomas in children. Neurosurgery 1991; 29: 14–18.
9. Brackmann DE, Kwartler JA. A review of acoustic tumors: 1983–1988. Am J Otol 1990; 11: 216–32.
10. Ojemann RG, Martuza RL. Acoustic neuroma, In: Youmans J R (ed) Neurological Surgery, 3rd edn. Philadelphia: WB Saunders; 1990, 3316–50.
11. Lunsford LD, Kamerer DB, Flickinger JC. Stereotactic radiosurgery for acoustic neuromas. Arch Otolaryngol Head Neck Surg 1990; 116: 907–909.
12. Rouleau GA, Merel R Lutchman M, et al. Alteration in a new gene encoding a putative membrane-organizing protein causes neuro fibromatosis type 2. Nature 1993; 363: 515–21.
13. Trofatter JA, MacCollin MM, Rutter JL, et al. A novel moesin-, ezrin-, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 1993; 72: 791–800.
14. Tsukita S, Yonemura S, Tsukita S. ERM proteins: head-to-tail regulation of actin-plasma membrane interaction. Trends Biochem Sci 1997; 22: 53–58.
15. McClatchey AI, Saotome I, Ramesh V, et al. The NF2 tumor suppressor gene product is essential for extraembryonic development immediately prior to gastrulation. Genes Dev 1997; 11: 1253–65.
16. Morrison H, Sherman LS, Legg J, et al. The NF2 tumor suppressor gene product, merlin, mediates contact inhibition of growth through interactions with CD44. Genes Dev 2001; 15: 968–80.
17. Rong R, Surace EI, Haipek CA, et al. Serine 518 phosphorylation modulates merlin intramolecular association and binding to critical effectors important for NF2 growth suppression. Oncogene 2004; 23: 8447–54.
18. Kim H, Kwak NJ, Lee JY, et al. Merlin neutralizes the inhibitory effect of Mdm2 on p53. J Biol Chem 2004: 279: 7812–18.
19. Lane DP, Crawford LV. T antigen is bound to a host protein in SV40-transformed cells. Nature 1979; 278: 261–63.
20. Linzer DI, Maltzman W, Levine AJ. The SV40 A gene product is required for the production of a 54,000 MW cellular tumor antigen. Virology 1979;98: 308–18.
21. Sarnow P, Ho YS, Williams J, Levine AJ. Adenovirus E1b-58kd tumor antigen and SV40 large tumor antigen are physically associated with the same 54 kd cellular protein in transformed cells. Cell 1982; 28:387–94.
22. Lane DP, Benchimol S. p53: oncogene or anti-oncogene? Genes Dev 1990;4: 1–8.
23. Weinberg RA. Positive and negative controls on cell growth. Biochemistry 1989; 28: 8263–69.
24. Mowat M, Cheng A, Kimura N, et al. Rearrangements of the cellular p53 gene in erythroleukaemic cells transformed by Friend virus. Nature 1985; 314: 633–36.
25. Wolf D, Rotter V. Major deletions in the gene encoding the p53 tumor antigen cause lack of p53 expression in HL-60 cells. Proc Natl Acad Sci USA 1985; 82: 790–94.
26. Masuda H, Miller C, Koeffler HR et al. Rearrangement of the p53 gene in human osteogenic sarcomas. Proc Natl Acad Sci USA 1987; 84: 7716–19.
27. Hinds R Finlay C, Levine AJ. Mutation is required to activate the p53 gene for cooperation with the ras oncogene and transformation. J Virol 1989; 63: 739–46.
28. Vogelstein B, Fearon ER, Hamilton SR, et al. Genetic alterations during colorectal-tumor development N Engl J Med 1988; 319: 525–32.
29. Hollstein M, Sidransky D, Vogelstein B, Harris CC. p53 mutations in human cancers. Science 1991; 253: 49–53.
30. Levine AJ, Momand J, Finlay CA. The p53 tumour suppressor gene. Nature 1991; 351: 453–56.
31. Malkin D, Li FP Strong LC, et al. Germ line p53 mutations in a familial syndrome of breast cancer, sarcomas, and other neoplasms. Science 1990; 250: 1233–38.
32. Avkin S, Sevilya Z, Toube L, et al. P53 and p21 regulate error-prone DNA repair to yield a lower mutation load. Molecular Cell 2006; 22: 407–13.
33. Benchimol S, Lamb P, Crawford LV, et al. Transformation associated p53 protein is encoded by a gene on human chromosome 17. Somat Cell Mol Genet 1985; 11: 505–10.
34. Lamb P, Crawford JL Characterization of the human p53 gene. Mol Cell Biol 1986; 6; 1379–85.
35. Unger T, Nau MM, Segal S, Minna JD. p53: a transdominant regulator of transcription whose function is ablated by mutations occurring in human career. EMBO J 1992; 11: 1383–90.
36. Horikoshi N, Usheva A, Chen J, et al. Two domains of p53 interact with the TATA-binding protein, and the adenovifus 13S E1A protein disrupts the association, relieving p53-mediated transcriptional repression. Mol Cell Biol 1995; 15:227–34.
37. Bargonetti J, Manfredi JJ, Chen X, et at. A proteolytic frapnent from the central region of p53 has marked sequence-specific DNA-binding activity when generated from wild-type but not from oncogenic mutant p53 protein. Genes Dev 1993; 7: 2565–74.
38. Pavletich NP, Chambers, KA, Pabo CO. The DNA-binding domain of p53 contains the four conserved regions and the major mutation hot spots. Genes Dev 1993; 7: 2556–64.
39. Kem SE, Kinzier KW, Bruskin A, et al. Identification of p53 as a sequence-specific DNA-binding protein. Science 1991; 252: 1708–11.
40. Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Crystal structure of a p53 tumor suppressor-DNA complex: understanding tumorigenic mutations. Science 1994; 265: 346–56.
41. Vogektein B, Kinzler KW. Tumour-suppressor genes. X-rays strike p53 again. Nature 1994; 370: 174–75.
42. Vogelstein B, Kinzler KW. p53 function and dysfunction. Cell 1992; 70: 523–26.
43. Kern SE, Pietenpol JA, Thiagalingam S, et al. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science 1992; 256: 827–30.
44. Farmer G, Bargonetti J, Zhu H, et al. Wild-type p53 activates transcription in vitro. Nature 1992; 358: 83–86.
45. Gannon JV Lane DP. Protein synthesis required to anchor a mutant p53 protein which is temperature-sensitive for nuclear transport. Nature 1991; 349: 802–806.
46. Momand J, Zambetti GP, Olson DC, et al. The Mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992; 69: 1237–45.
47. Haupt Y, Maya R, Kazaz A, Oren M. Mdm2 promotes the rapid degradation of p53. Nature 1997; 387: 296–99.
48. Honda R, Tanaka H, Yasuda H. Oncoprotein Mdm2 is a ubiquitin ligase E3 for tumor suppressor p53. FEBS Lett 1997; 420: 25–27.
49. Fang S, Jensen JP, Ludwig RL, et al. Mdm2 is a RING finger-dependent ubiquitin protein ligase for itself and p53. J Biol Chem 2000; 275: 8945–51.
50. Barak Y, Juven T, Haffner R, Oren M. Mdm2 expression is induced by wild type p53 activity. EMBO J 1993; 12: 461–68.
51. Carr AM. Cell cycle. Piecing together the p53 puzzle. Science 2000; 287: 1765–66
52. Sherr CJ, Weber JD. The ARF/p53 pathway. Curr Opin Genet Dev 2000; 10:94–99.
53. Meek DW. Mechanisms of switching on p53: a role for covalent modification? Oncogene 1999; 18: 7666–75.
54. Gatti A, Li HH, Traugh JA, Liu X. Phosphorylation of human p53 on Thr-55. Biochemistry 2000; 39: 9837–42.
55. Higashimoto Y, Saito S, Tong XH, et al. Human p53 is phosphorylated on serines 6 and 9 in response to DNA damage-inducing agents. J Biol Chem 2000; 275: 23199–203.
56. Canman CE, Lim DS, Cimprich KA, et al. Activation of the ATM kinase by ionizing radiation and phosphorylation of p53. Science 1998; 281: 1677–79.
57. Tibbetts RS, Brumbaugh KM, Williams JM, et al. A role for ATR in the DNA damage-induced phosphorylation of p53. Genes Dev 1999; 13: 152–157.
58. Jimenez GS, Khan SH, Stbmmel JM, Wahl GM. p53 regulation by post-translational modification and nuclear retention in response to diverse stresses. Oncogene 1999; 18: 7656–65.
59. Shieh SY, Ahn J, Tamai K, et al. The human homologs of checkpoint kinases Chk1 and Cds1 (Chk2) phosphorylate p53 at multiple DNA damage-inducible sites. Genes Dev 2000; 14: 289–300.
60. Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD. Phosphorylation of Ser-20 mediates stabilization of human p53 in response to DNA damage. Proc Nati Acad Sci USA 1999; 96: 13777–82.
61. Persons DL, Yazlovitskaya EM, Pelling JC. Effect of extracellular signal-regulated kinase on p53 accumulation in response to cisplatin. J Biol Chem 2000; 275: 35778–85.
62. Buschmann T, Potapova O, Bar-Shira A, et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol Cell Biol 2001; 21: 2743–54.
63. Biaydes JP, Vojtesek B, Bloomberg GB, Hupp TR. The development and use of phospho-specific antibodies to study protein phosphorylation. Methods Mol Biol 2000; 99: 177–89.
64. Kapoor M, Lozano G. Functional activation of p53 via phosphorylation following DNA damage by UV but not gamma radiation. Proc Natl Acad Sci USA 1998; 95: 2834–37.
65. Cuddihy AR, Wong AH, Tarn NW, et al. The double-stranded RNA activated protein kinase PKR physically associates-with the tumor suppressor p53 protein and phosphorylates human p53 on serine 392 in vitro. Oncogene 1999; 18: 2690–702.
66. Lu H, Taya Y, Ikeda M, et al. Ultraviolet radiation, but not ? radiation or etoposide-induced DNA damage, results in the phosphorylation of the murine p53 protein at serine-389. Proc Natl Acad Sci USA 1998; 95: 6399–402.
67. Sakaguchi K, Herrera JE, Saito S, et al. DNA damage activates p53 through a phosphorylation-acetylation cascade. Genes Dev 1998; 12: 2831–41.
68. Polyak K, Waldman T, He TC, et al. Genetic determinants of p53-induced apoptosis and growth arrest. Genes Dev 1996; 10: 1945–52.
69. Levy N, Yonish-Rouach E, Oren M, Kimchi A. Complementation by wild-type p53 of interleukin-6 effects on M1 cells: induction of cell cycle exit and cooperativity with c-myc suppression. Mol Cell Biol 1993; 13: 7942–52.
70. Sabbatini P, Lin J, Levine AJ, White E. Essential role for p53-mediated transcription in E1A-induced apoptosis. Genes Dev 1995; 9: 2184–92.
71. Vousden KH, Lu X. Live or let die: the cell's response to p53. Nat Rev Cancer 2002; 2: 594–604.
72. Haupt S, Berger M, Goldberg Z, Haupt Y. Apoptosis - the p53 network. J Cell Sci 2003; 116: 4077–85.
73. Wahl GM, Linke SP Paulson TG, Huang LC. Maintaining genetic stability through TP53 mediated checkpoint control. Cancer Surv 1997; 29: 183–219.
74. Kastan MB, Onyekwere O, Sidransky D, et al. Participation of p53 protein in the cellular response to DNA damage. Cancer Res 1991; 51: 6304–11.
75. be Laat WL, Jaspers NG, Hoeijmakers JH. Molecular mechanism of nucleotide excision repair. Genes Dev 1999; 13: 768–85.
76. Hanawalt PC. Subpathways of nucleotide excision repair and their regulation. Oncogene 2002; 21: 8949–56.
77. Tan T, Chu G. p53 binds and activates the xeroderma pigmentosum DDB2 gene in humans but not mice. Mol Cell Biol 2002; 22: 3247–54.
78. Fitch ME, Cross IV, Turner SJ, et al. The DDB2 nucleotide excision repair gene product p48 enhances global genomic repair in p53 deficient human fibroblasts. DNA Repair (Amst) 2003; 2: 819–26.
79. Therrien JP, Drouin R, Baril C, Drobetsky EA. Human cells compromised for p53 function exhibit defective global and transcription-coupled nucleotide excision repair, whereas cells compromised for pRb function are defective only in global repair. Proc Natl Acad Sci USA 1999; 96: 15038–43.
80. Smith ML, Seo YR. p53 regulation of DNA excision repair pathways. Mutagenesis 2002; 17: 149–56.
81. Fukasawa K, Choi T, R, et al. Abnormal centrosome amplification in the absence of p53. Science 1996; 271: 1744–47.
82. El-Deiry WS. Regulation of p53 downstream genes. Semin Cancer Biol 1998;8: 345–57.
83. Sah VP, Attardi LD, Mulligan GJ, et al. A subset of p53-deficient embryos exhibit exencephaly. Nat Genet 1995; 10: 175–80.
84. Johnson P, Chung S, Benchimol S. Growth suppression of Friend virus-transformed erythroleukemia cells by p53 protein is accompanied by hemoglobin production and is sensitive to erythropoietin. Mol Cell Biol 1993; 13: 1456–63.
85. Harvey M, Vogel H, Lee EY et al. Mice deficient in both p53 and Rb develop tumors primarily of endocrine origin. Cancer Res 1995; 55: 1146–51.
86. Gope ML, Chun M, Gope R. Comparative study of the expression of Rb and p53 genes in human colorectal cancers, colon carcinoma cell lines and synchronized human fibroblasts. Mol Cell Biochem 1991, 107: 55–63.
87. Bosari S, Viale G, Roncalli M, et al. p53 gene mutations, p53 protein accumulation and compartmentalization in colorectal carcinoma. Am J Pathol 1995, 147: 790–98.
88. O'Farreli TJ, Ghosh P, Dobashi N, et al. Comparison of the effect of mutant and wild-type p53 on global gene expression. Cancer Res 2004, 64: 8199–207.
89. Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997, 88: 323–31.
90. Moll UM, LaQuaglia M, Benard J, Riou G. Wild-type p53 protein undergoes cytoplasmic sequestration in undifferentiated neuroblastomas but not in differentiated tumors. Proc Natl Acad Sci USA 1995; 92: 4407–11.
91. Dittmer D, Pati S, Zambetti G, et al. Gain of function mutations in p53. Nat Genet 1993; 4: 42–46.
92. Louis DN. The p53 gene and protein in human brain tumors. J Neuropathol Exp Neurol 1994; 53: 11–21.
93. Lubbe J, von Ammon K, Watanabe K, et al. Familial brain tumour syndrome associated with a p53 germ line deletion of codon 236. Brain Pathol 1995; 5: 15–23.
94. Loeb LA. Mutator phenotype may be required for multistage carcinogenesis. Cancer Res 2001; 51: 3075–79.
95. Monoh K, Ishikawa K, Yasui N, et al. p53 tumor suppressor gene in acoustic neuromas. Acta Otolaryngol 1998; 537 Suppl : 11–5.
96. Dayaian AHPP Mathivanan J, Rohini K, et al. Age dependent phosphorylation and deregulation of p53 human vestibular schwannomas. Mol Carcinogenesis 2006; 45: 38–46.
97. Yamauchi M, Suzuki K, Kodama S, Watanabe M. Abnormal stability of wild-type p53 protein in a human lung carcinoma cell line. Biochem Biophys Res Commun 2005; 330: 483–88.
98. Thomas R, Prabhu PD, Mathivanan J, et al. Altered altered structure and expression of RB1 gene and increased phosphorylation of pRb in human vestibular schwannomas. Mol Cell Biochem 2005; 271: 113–21.
99. Xiao GH, Gallagher R, Shetler J, et al. The NF2 tumor suppressor gene product, merlin, inhibits ceil proliferation and cell cycle progression by repressing cyclin D1 expression. Mol Ceil Biol 2005; 25: 2384–94.
(c) Annals of Neurosciences.All Rights Reserved