Annals of Neurosciences, Vol 13, No 4 (2006)
Annals of Neurosciences, Volume 13, Issue 4 (October), 2006
THE TUMOR SUPPRESSOR GENE RETINOBLASTOMA (RB1) IN HUMAN VESTIBULAR SCHWANNOMAS
Corresponding author
Dr. Rajalakshmi Gope
Additional Professor
Department of Human Genetics
National Institute of Mental Health andNeurosciences
(NIMHANS) Bangalore 560 029.
Phone: 091 80 2699 5125 Fax: 091 80 2656 4830
Email:
Abstract
Tumorigenesis is a multistep process where normal control of cell proliferation and cell-cell interactions are lost and this leads to transformation of normal cells into tumor cell. Tumorigenic process involves atleast two classes of genes: (a) Oncogenes and (b) tumor suppressor (TS) genes. Activated oncogenes are known to aid uncontrolled cell growth whereas TS genes inhibit cell proliferation. The human retinoblastoma gene, RB1, is the first tumor suppressor gene to be identified. Vestibular schwannomas (VS) are one of the most common tumors of human central nervous system. Possible role of RB1 gene in human VS tumors are discussed here.
Key words: Tumor suppressor gene, RB1 gene, nervous system tumors, human vestibular schwannomas, NF2,
Human Vestibular Schwannomas
Human vestibular schwannomas (VS), also called as acoustic schwannomas account for approximately 8% of all primary intracranial tumors. Majority of these are sporadic (95%) and unilateral and present in the fourth and fifth decade. Anatomical location of these tumors makes management difficult and patients suffer great morbidity (1,2).
Symptoms of Vestibular Schwannomas
The clinical presentation of patients with VS can vary widely, but the earliest symptom is usually hearing loss, may be sudden or gradual, complete or incomplete. The typical tumor progression is associated with a gradual reduction in hearing. Other symptoms associated with VS include tinnitus, facial paralysis, vertigo, loss of balance and dizziness (3).
Tumor Diagnosis
Because unilateral hearing loss is the initial presentation for most patients with VS tumors, evaluation begins with routine audiologic testing using tests that include both pure tone and speech discrimination components. Approximately 5% of patients with unilateral sensorineural hearing loss have been found to harbour VS tumor (3). Computerized tomography (CT), enhanced with intravenous dye (contrast) is also critical in the early diagnosis of the tumor. The most reliable test is contrast-enhanced MRI in which the tumors appear as regular, brightly enhancing masses (
Epidemology
Prevalence
The incidence of VS appears to be greatest in the far Eastern part of the world, where they account for 10.6% of primary intracranial neoplasms in India and 10.2% in China (5, 6). Rates of 4.9% and 4% are reported in England and USA (7, 8) respectively. The lowest incidence is found in African Blacks, figures of 2.6% being recorded in Kenya, 0.9% in Nigeria, and 0.5% in what was formerly Rhodesia (9,10). The latter can be contrasted with a 3.7% incidence in the white population of the same country (11). A recent study in Denmark reported an incidence rate of 17.4 VS per one million inhabitants per year during the 1996–2001 which is almost double that reported during the period 1990–1995 (12). A study conducted over a period of 26 years has shown that the number of cases of human VS is increasing. However, the size and the age of onset of these tumors have been almost unchanged throughout the 26-year periodi 13).
Age and Gender Distribution
Vestibular schwannomas occur most frequently in middle age. In a series of 113 cases reported by the House et al., approximately 50% of patients were in their fifth or sixth decades, and only 15% patients are under the age of 30 (14). Vestibular schwannomas are rare in children, except in those who have neurofibromatosis type 2 (15). Cumulative results from large series in the literature show a preponderance of tumors in women (57%), as compared to men (43%). However, this is not true for childhood VS tumors where an equal sex distribution is seen (3,12).
Pathology
Gross morphologic features
Most VS tumors are lobular in form, well encapsulated, and solid. These tumors have a firm consistency, but there may be areas of soft tissue, and in some a cyst within the tumor may be found. The number of tumors arising from the superior and the inferior vestibular nerves are almost equal (16).
Histopathologic features
The classic description of schwannoma is a tissue composed of densely packed elongated spindle cells. Two tissue types, Antoni A and Antoni B constitute these tumors; Antoni B fibres are loose, semipalisading configurations of Schwann cells. Antoni A fibres are denser, with more numerous nuclei and substantially firmer cytoplasm. Other findings may include Verocay bodies, hemosiderine position, hyalinized blood vessels, recent and old thromboses, sheets of foamy macrophages, foci of high cellularity, whorls and collagenous scarring (17).
Management Strategies in Vestibular Schwannoma
The management of VS tumors is complex and multifaceted because of the high morbidity following surgery. The last century has witnessed effective management of VS where the mortality rates reduced from 50% to 11% (18). While monitoring useful facial and audiological function very small tumors are also closely followed without surgical intervention. Observation with serial monitoring by magnetic resonance imaging (MRI) at least every six months is an appropriate approach for patients with small and asymptomatic lesions. This permits the patient to enjoy a period of hearing as some of these tumors are slow-growing (14). As an alternative to conventional surgical techniques, radiosurgery (that is, radiation therapy – "the gamma knife" or LINAC) may be used to reduce the size or limit the growth of the tumor.
Genetic Factors
Vestibular schwannomas occur in both sporadic and familial forms. Unilateral sporadic tumors account for 95% of cases. Familial vestibular schwannomas result from NF2 (also known as neurofibromatosis type 2, bilateral acoustic neurinomas or central neurofibromatosis). It is an autosomal dominant condition with a high degree of penetrance. Development of tumors in NF2 patients follows the Knudson's "two-hit" hypothesis for tumor development (19). In NF2 patients carrying a germline mutation in one NF2 allele, a second hit in the other allele suffices to tumorigenesis. This concept is also supported by the fact that in sporadic VS both hits arise spontaneously within the same somatic cell (20,21).
Neurofibromatosis Type 2
Human NF2 tumors
NF2 is a dominantly inherited disorder and is one of the most common genetic disorders of the human nervous system. It has a population incidence of 1 in 30,000 to 40,000. NF2 is also characterized by a predisposition to the development of other tumors of the central and peripheral nervous system including meningiomas, ependymomas etc. (22). Another distinct clinical characteristic of NF2 patients is the development of sub-capsular cataracts in majority of the cases and appearance of café-au-lait spots in some. Mean age at the onset of symptoms for NF2 patients has been estimated to be approximately 22 years and at diagnosis, 28 years. Mean survival after diagnosis has been estimated to be 15 years.
According to NIH guidelines, diagnosis of NF2 is made in a patient who has: (1) Bilateral vestibular schwannomas OR family history of NF2 PLUS unilateral vestibular schwannomas OR any two of the following tumors such as: meningioma, glioma, neurofibroma, schwannoma, posterior subcapsular lenticular opacities. (2) Diagnosis of NF2 can also be made in a patient who has: unilateral vestibular schwannoma PLUS any two of the following tumors: meningioma, schwannoma, neurofibroma, posterior subcapsular lenticular opacities OR multiple meningiomas (2 or more) PLUS unilateral vestibular schwannoma OR any two of: glioma, neurofibroma, schwanoma, cataract. Occasionally, the disorder may occur as a spontaneous mutation with no family history. Some patients may not meet these criteria, but NF2 must be suspected in any patient below the age of 30 presenting with an acoustic neurinoma, any young patient with a Schwann cell tumor, and patients with multiple meningiomas (23).
NF2 gene and function of its protein
In 1986 and 1987, loss of heterozygosity (LOH) and cytological studies in NF2 cases has suggested the presence of a tumor suppressor gene on chromosome 22 (24). This hypothesis was confirmed when molecular genetic analysis of a large NF2 pedigree demonstrated linkage of NF2 to chromosome 22q 12. Additional linkage studies narrowed down the location of the NF2 gene, and in 1993 two groups cloned the gene independently (20,21). The NF2 gene spans 110 kilobases and comprises 16 constitutive exons and one alternatively spliced exon. The NF2 protein exists in two major alternative forms. Isoform 1 consists of 595 amino acids produced fromexon 1 through 15 and exon 17. Isoform 2 contains alternatively spliced protein where the exon 16 is included. As a result there is alteration at the C terminus of the protein replacing 16 amino acids with 11 novel residues. The NF2 gene encodes a cytoskeletal protein, merlin or schwannomin that appears to have a rple in modulating cellular motility and proliferation (20,21). Merlin is the first human tumor suppressor proposed to function through structural links at the plasma membrane and the actin cytoskeleton. Inactivation of the NF2 gene and consequent lack of gene expression appears to be the primary cause of the disease (25). Merlin is highly expressed in the nervous system but is nonetheless present in a surprisingly limited number of cell types. Merlin is expressed in Schwann cells, melanocytes, red blood cells and endothelial cells. Reports show NF2 gene defects in non-NF2 tumors like mesotheliomas, thyroid carcinomas, hepatocellular carcinoma cell lines and perineurial tumors (26). Mutations of the NF2 gene in tumors from familial and sporadic cases and some tumors unrelated to NF2 indicate that the NF2 gene may constitute a classic tumor suppressor gene (27).
Targetted inactivation of NF2 gene in the Schwann cells of mice developed schwannomas and Schwann cell hyperplsia (28). Heterozygous NF2 mutant mice spontaneously developed a wide range of highly metastatic tumors including osteosacromas at high frequency, and fibrosarcoma, and hepatocellular carcinoma at a lower rate (29). Overexpression of merlin in oncogene-transformed cell lines reverted the oncogene-induced phenotype and overexpression in NIH3T3 cells or RT4 and JS1 rat schwannoma cell lines led to reduced proliferation (30–32).
Suppression of merlin synthesis in tumor cell lines increased proliferation. Overexpression of the normal but not mutant NF2 gene inhibited actin-cytoskeleton-mediated processes including cell motility, cell spreading and cell attachment (33). The control of Schwann cell proliferation is lost by the inactivation of NF2 gene, which suggests that merlin deficiency disrupts some aspect of intracellular signaling that leads to cellular proliferation (34).
On the basis of sequence similarity, merlin was determined to be a member of the protein 4.1 family of cytoskeleton-associated proteins. The ERM proteins (ezrin, moesin, radixin), to which the NF2 protein is most closely related, share 70–75% amino acid identity with each other. Like the ERM proteins, merlin is predominantly found in the membrane ruffles and cellular protrusions (35). Eventhough there is high homology in the amino-terminal region between ERM proteins and merlin, differences exist in the carboxy-terminal domain, suggesting that merlin has functions distinct from other ERM proteins (36) (
Under certain conditions merlin was found in the nucleus in cultured cells (39). However, the sum of available data is consistent with the notion that merlin normally carries out its growth-suppressing activity from the cell periphery; perhaps merlin is removed from the periphery and sequestered in the nucleus during conditions of exponential proliferation (40).
Structure of NF2 protein
All the members of the ERM family of proteins, which include ezrin, radixin moesin, have the similar structural organization: a large globular N-terminus domain, a long alpha-helical segment and a small charged C-terminus region (20,21). The N-terminal FERM (4.1, ezrin, radixin and moesin) domain is made of three sub-domains designated A to C (
The Rretinoblastoma (RB1) Gene
The identification of tumor suppressor genes represents a crucial milestone in the understanding of cancer genetics (41). The products of tumor suppressor genes are involved in a variety of cellular functions. The archetypal tumor suppressor is frequently represented by the retinoblastoma gene and loss of its function is responsible for susceptibility to retinoblastoma, a sporadic or familial, pediatric neoplasm arising from retinal cells harboring either delete or mutational inactivation of both RB1 alleles (19). RB1 complies with all of the requirements to be biologically defined as a bona fide tumor suppressor gene (42).
First, it was experimentaly demonstrated that re-introduction of wild-type RB1 allele into cells derived from RB1-deficient retinoblastomas and many other human tumors suppressed neoplstic phenotypes like anchorage-independent growth and tumorigenesis in nude mice. With the development of transgenic mouse and gene targeting techniques, the concept that RBI inactivation led to tumorigenesis was recapitulated (43,44). Mice deficient in RB1 gene exhibit embryonic lethality, dying between embryonic days E13.5 and E15.5, and suffer from abnormal differentiation in a variety of tissues, most probably due to inappropriate cell-cycle entry and elevated apoptosis. Inactivation of RB1 gene results in aberrant cell-cycle, which is characterized by inadequate response to the extracellular growth-inhibitory signals (45). Mutations affecting the retinoblastoma gene are frequently encountered in retinoblastomas and in other cancers like osteosarcoma, soft-tissue sarcoma, small cell lung cancer, bladder cancer, prostrate cancer, and breast cancer (41,45–47). It is clear that the protein product of RB1 gene, pRb, has multiple cellular functions and resides in multiple protein complexes. Accumulating evidence suggests that pRb is critical for growth signals, differentiation and embryonic development (41,45,48).
RB1 Gene Structure and Localization
The human RB1 gene is localized on chromosome 13ql4. The gene consists of 27 exons dispersed over 200 kb which encodes a 4.7 kb mRNA and 110 kDa pRb protein with 928 amino acids. The exons range from 32 to 1,889 base pairs (bp) in length and introns range 80 bp to 60 kb (46,50). Several features of the RBI gene promoter are reminiscent of characteristics associated with many "housekeeping" genes, consistent with its ubiquitous expression pattern (50). High-resolution deconvolution microscopic studies have revealed that, during G1 and S phases, the three pocket proteins including pRb and the related proteins pl07 and pl30 are found in the perinucleolar foci (51).
Retinoblastoma Family Members
pRb is the prototype of a family of proteins, referred to as the pocket proteins, that includes pl07 and pl30 (52). pRb shares significant structural homology with pl07 and pl30, particularly in the "pocket region," hence referred to as pocket proteins. To study the physiological role of each pocket protein, mice lacking; pRb, pl07 and pl30 were developed. Mice deficient in RB1 gene exhibit embryonic lethality, dying between embryonic days E13.5 and E15.5, and suffer from abnormal differentiation in a variety of tissues, perhaps due to inappropriate cell-cycle entry and increased apoptosis. There is significant functional overlap between the pocket proteins (53). The pocket proteins can interact with the same set of viral oncoproteins and a subset of the same cellular gene products that interact with pRb. For example, all of the pocket proteins can bind the E2F family of transcription factors, cyclins, and histone deacetylases. All of the pocket proteins can be phosphorylated and regulated by cyclin dependent kinases (CDKs).
Mutation of RB1 gene is observed at a detectable frequency in almost all types of human cancers. In contrast, mutation of pl07 has only been detected in a B cell lymphoma cell line (54). Mutation or alteration of RB2/pl30 expression is detected more frequently in cancer than mutation or alteration of pl07 expression. Inactivating mutations of RB2/pl30 have been discovered in lung cancer (55) and Burkitt's lymphoma (56). pRb interacts predominantly with E2Fs 1,2,3 and 4, whereas pl07 and pl30 primarily interact with E2F4 and E2F5 (53). While pRb interacts with E2Fs in both cycling and quiescent cells, pl30 interacts with E2Fs primarily in quiescent cells. However, pl07 is prominently associated with E2Fs in the S phase of the cell cycle (57). In addition, pl07 and pl30 can recruit CDK-2 containing complexes to promoters, and some experiments have indicated that pl07 and pl30 can act as inhibitors of CDK function.
pRb Protein Structure
The RB1 gene encodes a relatively stable nuclear phospho-protein, pRb (half-life >8 h) of 928 amino acids. Although there are differences in the levels of pRb in different cell types, the protein is widely and constitutively expressed. The structure of the RB1 promoter, being GC-rich and lacking canonical TATA or CAAT boxes, is similar to other constitutively expressed "house-keeping" genes. Although the level of pRb is relatively constant in many cell types, its function is modulated by post-translational modification. This post-translational regulation consists primarily of serine/threonine phosphorylation that occurs in synchrony with the cell cycle (58–61) (
Purified pRb contains three structural domains that are protease resistant in vitro: the N terminal region, the central A and B domains separated by a linker region, and the C-terminal region. The structural integrity of the A/B domains is required for the interaction of pRb with most of its associated proteins. The N domain (beginning at amino acid 8) is 40 kDa in size and constitutes most of the amino-terminal half of pRb. Continued proteolysis of the N domain generates two sub-fragments of 30 kDa (beginning at amino acid 8) and 10 kDa (beginning at amino acid 263). Although N domain sequences are required for interaction with some cellular proteins, the contribution this domain makes to normal pRb function is unclear. This region contains consensus cdk phosphorylation sites, which may regulate pRb activity when they are phosphorylated during the cell cycle (
Domains A and B are highly conserved from humans to plants, and they interact with each other along an extended inter-domain interphase to form the central pocket, which is critical to the tumor suppressor function of pRb (62). The A domain (beginning at amino acid 377) and B (beginning at amino acid 622) domains have apparent molecular mass of approximately 24 and 20 kDa, respectively, and map to the carboxy terminal half of the protein. One of the first biologically relevant pRb activities uncovered was its ability to bind the oncogenic proteins, like adenovirus E1A and human papillomavirus E7, from the small DNA tumor viruses (63). The ability of these viral proteins to transform cells in vitro correlated with their ability to bind pRb suggesting that they may induce tumorigenesis, in part, by blocking pRb function. Deletion mapping studies indicted that the A (amino acids 379–572) and B domains (amino acids 646–772) are necessary for mediating interaction with these proteins. The viral onco-proteins bind pRb through an LXCXE amino acid motif. The A and B domains are sufficient for mediating interaction with cellular proteins, like histone deacetylases, that contain a similar LXCXE pRb-binding motif. A 74 amino acid spacer separates the A and B domains, the A domain, spacer, and B domain are collectively called the "small pocket". The importance of small pocket for pRb-mediated tumor suppression is underscored by the fact that many naturally occurring RB1 mutations disrupt its integrity. The A and B domains are the most conserved regions of pRb throughout evolution. Disruption of small pocket compromises pRb activity in most in vitro functional assays. Phosphorylation sites are also absent within the A/B domains suggesting that modification of this structure is not tolerated/Therefore, the integrity of the small pocket is essential for normal pRb function (
In addition to the small pocket, sequences carboxy-terminal to the B domain is critically important for pRb function. This C domain is sufficient to mediate interaction with a subset of pRb binding partners including Cdk2, PPla, UBF; MDM2, and c-abl (64). The tyrosine kinase activity of c-abl is blocked when it is complexed with pRb. This interaction appears to be important for pRb-mediated growth suppression. When pRb is hyper-phosphorylated, active c-abl is released. The importance of pRb-MDM2 interaction is less clear.
Functions of pRb
pRb regulates G1 phase of the cell-cycle
A key component of the machinery that regulates cell-cycle entry and progression in mammalian cells is the retinoblastoma protein (pRb), which functions as a barrier to inappropriate cell-cycle progression (65). A role for tumor suppressors in cell-cycle regulation is exemplified by studies of the gene product of RB1. Over expression of RB1 gene by microinjection into cells of purified unphosphorylated pRb protein in early G1 phase results in reversible G1 arrest, while injection of similar amounts of pRb in late G1 phase or early S phase has no effect on DNA synthesis (66). Similar results are observed in cells overexpressing RBI gene through transfection of an exogenous RB1 cDNA (67). Thus, pRb controls a restriction point that is controlled by pRb during the cell cycle transition from early G1 phase to late G1/S phase. Passage through this point commits normal cells to DNA synthesis and cell division. pRb mostly regulates G1 to S phase transition by its interaction with the E2F family of transcription factors (
E2F Family of Proteins
The growth suppressive properties of pRb are thought to be largely dependent upon its ability to regulate the E2F transcription factors. E2Fs control the expression of genes that are essential for cell proliferation, including key components of both the DNA-replication and cell-cycle control machinery. pRb binds to E2F during the G1 phase of the cell-cycle and inhibits the activation of its target genes (66). Dissociation of the pRb-E2F complex is dependent upon the sequential phosphorylation of pRb by cyclinD/cdk4andcyclinE/cdk2 (
Other mechanisms of transcriptional repression by pRb
pRb exerts its function by repressing the transcription of S-phase specific genes preventing transition to S-phase when cells are not in the cycling phase. pRb regulates this by its interaction with the E2F family of transcription factors. pRb is associated with several proteins other than E2F in transcriptional repression. The association of pRb with histone deacetylases HDAC1, HDAC2, and. HDAC3- has been established (69). pRb-mediated transcriptional repression involves alteration of higher order chromatin structure, which is regulated by nudeosome re-modeling complexes in an ATP-dependant manner (70).
Regulation of pRb Function by Phosphorylation
The phosphorylation status of pRb is a decisive factor in its role in cell-cycle. Cell cycle-dependant phosphorylation of pRb in vivo was first reported in 1989 (58–61,71). It was shown that phosphorylation begins in late Gl and continues until M phase (58–61,65). The hypophosphorylated form of pRb is active during growth suppression and hyperphosphorylated forms are inactive. pRb is phosphorylated by cyclin-dependant kinases (CDKs)in vitro (60,67,71,72). pRb contains 16 potential Ser/Thr sites for cdk phosphorylation, and it oscillates between hypophosphorylated and hyperphosphorylated forms during the cell-cycle. Atleast three different cyclin-cdk complexes have been suggested to phosphorylate pRb during the cell cycle. It is thought that cyclin D-cdk4/6 phosphorylates pRb in early G1, cyclin E-cdk2 -phosphorylates the protein near the end of G1, and cyclin A-cdk2 may maintain phosphorylation of pRb during S-phase (68). pRb phosphorylation is initiated in a growth factor-dependent manner by assembly of cyclin D with CDK4. Phosphorylation is then accelerated during late G1 by cyclin E-CDK2. Maintenance of the phosphorylated state during S and G2 is due to the actions of cyclin A-and cyclin B-CDK complexes. Cyclin-CDK complexes phosphorylate multiple proline-directed consensus sites on pRb (
Phosphorylation of specific sites appears to regulate distinct pRb functions, suggesting complex regulation of pRb by these phosphorylation events (74). Starting in the late G1 phase of the cell-cycle, pRb is heavily phosphorylated until mitosis. Hypophosphorylated pRb is the active form of pRb that negatively regulates E2F and cell-cycle entry. The D-type cyclin-CDKs are highly responsive to growth factor stimulation at several levels* These include synthesis of its subunits, association with inhibitory proteins such as cyclin kinase inhibitors (CKIs), assembly of the subunits, and cyclin stability (75). pRb is reactivated by hypophosphorylation at the end of mitosis (
Mechanisms of RB1 Gene Inactivation
Retinoblastoma protein (pRb) contains multiple protein binding sites and functions as a molecular scaffold to promote the assembly of transcription factors. Disassembly of these complexes is mediated by pRb inactivation by means of four known mechanisms, (a) The RB1 gene is mutated, causing release of its associated factors. RB1 mutations have been detected in retinoblastomas and a small fraction of sporadic tumors (41,45,47). (b) RB1 is sequestered by viral oncoproteins, such as E1A, which prevent it from binding other factors (59). (c) Phosphorylation of pRb by CDK-cyclin complexes during cell-cycle progression disrupts its ability to assemble transcriptional complexes (68). (d) pRb is degraded by caspase-dependent proteolytic pathway during apoptosis (87).
Role of pRb Beyond G1 Phase
In addition to its role in G1-S phase transition, pRb is also known to have some contribution in the later stages of cell-cycle. The presence of the Rb-hSW1/SNF complex in S phase and continued repression of the cyclin A and cdc2 genes suggest a role for pRb in the remote control of S phase exit and subsequent M phase entry. Therefore, pRb not only plays a role in controlling the cell cycle passage through the Gl restriction point, but also coordinates the regulatory machinery in G1 with that in S, G2 and M phase by orchestrating a cascade of cyclin induction and cdk activation (68).
pRb in Differentiation
pRb is known to function in the regulation of terminal differentiation of many tissue types (48,77,78). pRb knockout mice exhibit developmental defects in neurogenesis and erythropoiesis (43,44). It has been demonstrated that these developmental defects caused by loss of function of pRb during differentiation.
Role of pRb in Neuronal Differentiation
Many reports show a role of pRb during differentiation of nervous system. In the RB1-/- mice the central and peripheral nervous system (CNS/PNS) phenotype initiates at that time during mouse embryo development when neuronal precursor cells normally exit from the cell cycle and begin neuronal differentiation, suggesting that pRb function might be specifically required in the process of neurogenesis (79).
RB1+/- mice develop pituitary tumors rather than retinoblastoma (43), although chimeric mice comprising a mixture of wild-type cells and cells lacking both RBI gene and the other related protein pl07 do develop retinoblastoma. Another finding was that deletion of RB1 gene in the embryo itself, and not in the placenta, did not show the pronounced cell death in the central nervous system that was previously observed in RB1-/- mice (80). So, the apoptosis originally observed in RB1-/- neurons was not a cell-intrinsic defect but a secondary effect of placental dysfunction. Studies using inactivation of RB1 gene in specific cell populations in the nervous system showed that RB1-deficient neurons undergoectopic S-phase entry, but do not show defects in neuronal apoptosis or differentiation (81). Another report suggests that retinoblastoma gene promoter directs trans-gene expression exclusively to the nervous system (82). There is evidence that RB1 gene regulation of differentiation and apoptosis in the nervous system is highly specific. Selective inactivation of RB1 gene in the developing retina showed that this gene has distinct cell-autonomous roles at different stages of development, regulating proliferation of progenitor cells and rod photoreceptor differentiation in post mitotic cells. In the developing cerebellum, RB1 gene loss did not induce any anomalous proliferation or apoptosis in purkinje cells; however the neighbouring RB1-/-cerebellar granule neurons showed abnormal proliferation, disrupted differentiation and widespread apoptosis. By contrast, combined deletion of pRb and p53 in cerebellar granule neurons resulted in medulloblastoma (83), indicating pRb-deficient apoptosis is p53-dependent in these cells. These results suggest RB1 gene has important role in neurogenesis.
Role of pRb in the Maintenance of Genomic Stability
Susceptibility of the genome to multiple genetic alterations can be attributed to a loss of function in maintaining genomic stability (84). pRb is known to interact with two groups of proteins involved in chromosome segregation: (1) mitosin, astructural component of the kinetochore and (2) Heel, a conserved regulator of multiple mitotic events (85). Therefore, loss of pRb could disrupt the process of chromosome segregation leading to chromosomal instability.
pRb has been suggested to inhibit new DNA synthesis, that is, re-replication, before the completion of chromosome segregation and cytokinesis (86). In RB1-deficient cells multiple types of chromosomal aberrations have been observed since pRb appears to be modulating chromosome replication, segregation, and structural maintenance (86).
Regulation of Apoptosis by pRb
Several other lines of evidence suggest that pRb may also regulate apoptosis negatively (87). Previous reports from colon tumors, cultured colon tumor cell lines, breast cancer and bladder cancer indicated tumor specific increase in the level of pRb and the phosphorylated form -of pRb. A positive role for hyperphos-phorylated pRb in malignant transformation is also implied in human colon cancer (87–89) and bladder cancer.
Several results suggest that the E2Fs provide a good mediator for how pRb regulates both cell proliferation and apoptosis. Ectopic expression of E2F1, and other members of the E2F family, has been shown to induce apoptosis in tissue culture and in transgenic mice model (90).
Combined loss of pRb and p53 is known to be highly tumorigenic in mice (64,65,68). All these data point to the role of pRb as a negative regulator of apoptosis, which is contrary to its function as a tumor suppressor.
Regulation of Tumor Suppressor and Anti-Apoptotic Functions of pRb
pRb inhibits apoptosis, which seems to oppose its tumor suppressor activity. In tumors with functional RB1 gene the function of pRb is modulated by hypo and hyperphosphorylation (58–61). Most adult solid tumors have intact RB1 gene and protein and these are generally slow growing, resistant to apoptosis, and insensitive to chemotherapy and radiation. In contrast, tumors containing inactive RB1 gene such as retinoblastoma and small cell lung cancer usually have a high rate of proliferation and apoptosis and are generally sensitive to chemotherapy and radiation. Similarly, in cultured cells and mouse models, genetic inactivation of pRb leads to increased cell proliferation and apoptosis (43,44) whereas phosphorylation of pRb (e.g. as a result of pl6 INK4a inactivation) generally does not have this effect (91). A possible explanation for these findings is that the tumor suppressor functions of pRb may be regulated by variable phosphorylation events of pRb than the antiapoptotic activity. It is shown that the tumor suppressor and antiapoptotic functions of pRb are regulated by distinct phosphorylation events. As a survival strategy, some cancer cells may exploit this dual role of pRb by phosphorylating sites that regulate tumor suppression but voiding phosphorylation of Ser 567 and consequent apoptotic stimulus (92).
RB1 Gene Alterations in Human Tumors
The analysis of human tumors has revealed a wide spectrum of mutations that alter the RBI pathway (45–47, 49). Although mutation of the RB1 gene was first observed in inherited retinoblastomas, it is known that loss of pRb function contributes to a wide array of human cancers (68). RB1 gene is expressed in a number of human fetal tissues and organs and the level of expression is similar in all tissues examined (49). Alterations in RB1 expression have been reported in many human tumor types including lung cancer, osteosarcomas, leukemias, prostate cancer and bladder cancer (45–47, 93). Increased expression of RB1 mRNA has been reported for many human colon tumor tissues and human colorectal cancer cell lines and breast cancers (87–89).
RB1 Gene Alterations in Brain Tumors
RB1 tumor suppressor gene has been studied in several intracranial tumors including gliomas, meningiomas, pituitary adenomas etc. There are reports that loss of pRb expression in pituatory tumors (94) and glioblastomas (95) are associated with promoter hypermethylation. In gliomas, specific evidence for RB1 gene involvement was shown in approximately 13% of astrocytomas all of which were Grade 4. It was also demonstrated that the RB1 gene inctivation and loss of its protein (p110 Rb) expression might be associated with glial tumor progression.
RB1 Gene in Human Vestibular Schwannomas
LOH of RB1 gene was detected in 44% of malignant peripheral nerve sheath tumors (96). A recent study found that CDK2 was significantly downregulated in vestibular schwannomas and this could in turn deregulate the pRb-CDK pathway (97). But this study was limited to only eight VS samples and thus the data was insufficient to establish the possible role of RB1 gene in these tumors. Using cell line that lack NF2 protein merlin, it was show that the wild-type merlin represses cyclin D, in turn inhibits CDK4 activity leading to lower levels of phosphorylated form of pRb (98). Thus, the NF2 protein, merlin, is known to exert its tumor suppressor function through kinase that phosphorylates the pRb (98).
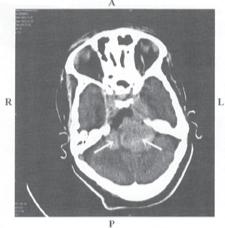
Figure 1: CT scan picture of bilateral vestibular schwannoma
Computerised tomography (CT) scan demonstrating bilateral vestibular schwannoma in ayoung female who had hearing loss and facial paralysis. The VS on the left was much larger than that on the right.
Surgery was done on the right side.
R-right; L-left; A-anterior; p-posterior
Image courtesy Department of Neuroimaging & Interventional Radiology, NMHANS Thomas R et al 2005 (ref 99), Dayalan AH et al 2006 (ref 100)
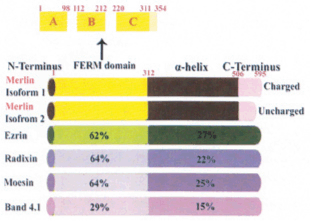
Figure 2: Merlin and the ERM proteins
Domain structure of merlin isoforms in relation to ezrin-radixin-moesin (ERM proteins).
The last 16 amino acids in isoform 1 are replaced by 11 novel residues in isoform 2. The N-terminal FERM domain is divided into 3 subdomains (A-C) which is shown here. The amino-acid identity of merlin isoform 1 to FERM family members and band 4.1, the superfamily is also shown. All four proteins are members of the band 4.1 superfamily and have a FERM (4.1, ezrin, radixin, moesin) domain – a 300 aminoacid module at the amino terminus that is involved in localizing to the plasma membrane and mediating interactions between the membrane and the cytoskeleton.
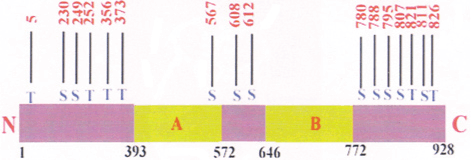
Figure 3: Phosphorylation sites of pRb
There are 16 potential serine/threonine phosphorylation sites in pRb and it oscillates between hypophosphorylated and hyperphosphrylated forms during the cell-cycle. At the G1-S phase transition, pRb is thought to be phosphorylated by CDK2, CDK4, and CDK6. hyperphosphrylated pRb releases E2F; allowing it to activate transcription of its target genes. pRb is reactivated by hyperphosphrylation at the end of mitosis.
S-Serine ; T – Threonine
The N and C terminus are shown. The A and B domains of pRb are also represented
The numbers 1 to 928 indicates the amino acids, of pRb.
Adapted from Less JA et al, `1991 (ref 73) Knudsen ES and Wang JY, 1996 Sherr CJ and Roberts JM, 1999 (ref 75), Ludlow JW and Nelson DA, 1995 (ref 76)
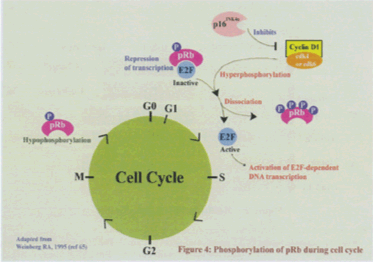
Figure 4: Phosphorylation of pRb during cell cycle
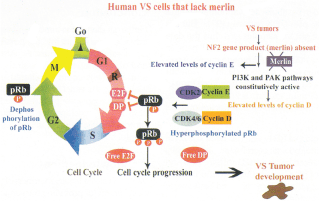
Figure 5: Role of merlin in cell-cycle; The pRb pathway is deregulated in the absence of NF2 protein, merlin.
This figure shows the scenario in the absence of wild-type merlin. pRb plays a very important role in cell-cycle especially at the G1-S checkpoint. The level of phosphorylated pRb is a very important regulator of the cell-cycle. All human VS lack the NF2 protein, merlin. In the absence of merlin there is increased level of phosphorylated pRb due to constitutively active form of cyclin D1-CDK4 complex and cyclin E-CDK2 and this could drive the VS cells towards cycling phase and tumor development. G0 – resting phase; G1 – pre-synthetic phase; S – Synthetic phase; G2 – post-synthetic phase, M-mitotic phase; R – restriction point Adapted from Xiao GH et al, 2005 (ref 98) Rong et al, 2004 (ref 38) Thomas R et al, 2005 (ref 99)
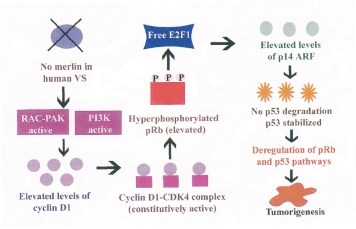
Figure 6: Possible pRb and p 53 pathways in vS tumor development ; Merlin exerts its tumor suppressor function via pRB and p 53.
During VS tumorigenesis, the pRb and the p53 pathways appear to be deregulated. Merlin is known to exert its tumor suppressor functions by inhibiting the p21-activated kinase (PAK) and the phosphatidyl inositol-3 kinase (PI3K) pathways. These two pathways converge on constitutive cyclin D1 activation and hence progression into cell-cycle. IN the absence of merlin in human Vs these pathways are constitutively active which results in elevated levels of cyclin D-CDK-4 complex and hence inaeased levels of phosphorylated pRb. The E2F1 transcription factor is free to induce the expression of pl4ARF which binds and sequesters MDM2 in the cucleolus, preventing MDM2-mediated export of p53 to the cytoplasm for degradation hence leading to p53 accumulation. Therefore, the deregulated p53 and pRb pathways could lead to uncontrolled proliferation and tumor formation.
Adapted from Xiao GH et al, 2005 (ref 98), Rong R et al, 2004 (ref 38) Themas R et al, 2005 (ref 99) Dayalan AH et al, 2006 (ref 100)
LOH in the non-coding region of RB1 gene has been shown in human VS tumors which is indicative of overall genomic instability in these tumors (99). Increased levels of RB1 mRNA, pRb and the phosphorylated form of pRb has also been found in these tumor tissues (99) indicating that RB1 gene could have major role in the NF2 tumor development.
The cyclin-CDK complex regulates the phosphorylation of pRb at the G1 to S- phase transition. Merlin is known to exert its tumor suppressor functions by inhibiting the p21-activated kinase (PAK) and the phosphatidyl inositol-3-kinase (PI3K) pathways. These two pathways converge on cyclin D1 activation, permitting the cells to progress through the cell-cycle. Therefore, in the absence of merlin in human VS tumors these pathways could be deregulated which could result in elevated levels of cyclin D1-CDK4 complex and this in turn could lead to increased levels of phosphorylated form of pRb (99). A recent finding further supports this hypothesis: the levels of cyclin E are regulated by wild type merlin through the “Hippo” signaling pathway. Therefore, in the absence of functional merlin in human VS tumors there is deregulated phosphorylation of pRb that is mediated by cyclin D-CDK4/6 and cyclin E/CDK2 and these changes in turn could lead to VS tumor development (99) (
The two fundamental molecular pathways, the pRb and p53 pathways, regulate cell growth, differentiation and cell death. The importance of these pathways in cellular growth control is underscored by the observation that members of these pathways are mutated in a majority of human cancers. It is conceivable that these two fundamental pathways could be deregulated in human VS tumors because of the absence of NF2 gene product, merlin in these tumors (
Conclusion
Further studies on the tumor suppressor genes RB1 and p53 could provide more insight into the possible role of these genes in the development of VS tumors. At present it appears that the kinases and phosphatases which control cell-cycle, cell differentiation and apoptosis could be altered in these tumors. Therefore, identifying the exact mechanism leading to the deregulation of the tumor suppressor pathways, perhaps kinase pathways, could help in the development of early diagnostic and treatment protocols which should inturn help us to better manage these painful human tumors.
Acknowledgement
We thank all the faculty members of the NIMHANS Neurosurgery department for providing the surgical tumor tissues which were used in some of the publications mentioned in this review. Part of the results discussed here are from the research project funded by the Department of Biotechnology (DBT), Government of India, to Dr. R.Gope, project number BT/PRO 703/MED/ 09/133/97. Financial assistance in the form of Junior and Senior Research Fellowships (JRF, SRF) from ICMR (AHPPD), CSIR (RT) and UGC (MJ and RK) are gratefully acknowledged. According to the journal format only a maximum of 100 references are cited in this review. Many more relevant reference could not be cited here and we applogise to the authors whose references are not included.
References
1. Cushing H. Intracranial Tumors. Springfield, IL: Ed., Charles C. Thomas, 1932.
2. Pool JL, Pava AA. eds. The early diagnosis and treatment of acoustic nerve tumors. 1st ed. Springfield, Illinois: Thomas, 1957; 22.
3. Ojemann RG. Acoustic neuroma (vestibular schwannoma). In: Kaye AH, Laws ER, eds. Brain tumors, 2nd ed. London: Churchill Livingston. 2001; 671–86.
4. Kocaoglu M, Bulakbasi N, Ucoz T, et al. Comparison of contrast-enhanced T1-weighted and 3D constructive interference in steady state images for predicting outcome after hearing-preservation surgery for vestibular schwannoma. Neuroradiology 2003; 45: 476–81.
5. Dastur DK, Lalitha VS, Prabhakar V. Pathological analysis of intracranial space-occupying lesions in 1000 cases including children. Age, Sex and pattern; and the tuberculomas. J Neurol Sci 1968; 6: 575–92.
6. Wen-qing H, Shi-ju Z, Qing-sheng T, et al. Statistical analysis of central nervous system tumors in China. J Neurosurg 1982; 56: 555–64.
7. Barker DJ, Weller RO, Garfield JS. Epidemiology of primary tumors of the brain and spinal cord: a regional survey in Southern England. J Neurol Neurosurg Psychiatry 1976; 39: 290–96.
8. Kurland LT, Schoenberg BS, Annegers JF, et al. The incidence of primary intracranial neoplasms in Rochester, Minnesota 1935–1977. Ann NY Acad Sci 1982; 381: 6–16.
9. Ruberti RF, Poppi M. Tumors of the central nervous system in the Africans East Afr Med J, 1971; 48: 576–84.
10. Adeloye A. Neoplasms of the brain in the African Surg Neurol 1979; 11: 247–55.
11. Levy LF; Auchterlonie WC. Primary cerebral neoplasms in Rhodesia Int Surg 1975; 60: 286–93.
12. Tos M, Stangerup SE, Caye-Thomasen P, et al. What is the real incidence of vestibular schwannoma? Arch Otolaryngol Head Neck Surg 2004; 130: 216–20.
13. Stangerup SE, Tos M, Caye-Thomasen P, et al. Increasing annual incidence of vestibular schwannomas and age at diagnosis. J Laryngol Otol 2004; 118: 622–27.
14. House JW, Nissen RL, Hitselberger WE. Acoustic tumor management in senior citizens. Laryngoscope 1987; 97: 129–30.
15. Allcut DA, Hoffman HJ, Isla A, et al. Acoustic schwannoma in children. Neurosurgery 1991; 29: 14–8.
16. Friedman RA, Kesser BW, Slattery WH, et al. Hearing preservation in patients with vestibular schwannomas with sudden sensorineural hearing loss. Otolaryngol Head Neck Surg 2001; 125: 544–51.
17. Sobel RA, Wang MD. Vestibular (acoustic schwannomas): Histologic features in neurofibromatosis 2 and in unilateral cases. J Neuropathol Exp Neurol 1993; 52: 106–13.
18. Gormiey WB, Sekhar LN, Wright DC, et al. Acoustic neuromas: results of current surgical management. Neurosurgery 1997; 41: 50–60.
19. Knudson AG, Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc Nat Acad Sci USA 1971; 68: 820–23.
20. Trofatter JA, MacCollin MM, Rutter JL, et al. A novel moesin-, ezrin, radixin-like gene is a candidate for the neurofibromatosis 2 tumor suppressor. Cell 1993; 72:791–800.
21. Rouleau GA, Merel P, Lutchman M, et al. Alternation in new gene encoding a putative membrane-organizing protein causes neurofibromatosis type 2. Nature 1993; 363: 515–21.
22. Martuza RL, Eldridge R. Neurofibromatosis 2 (bilateral acoustic neurofibromatosis). N Engl J Med 1988; 318: 684–88.
23. Martuza RL, Rouleau G. Genetic aspects of neurosurgical problems In: Youmans J R (ed.) Neurlogical Surgery, 3rd edn. Phildelphia; WB Saunders : 1990; 1061–80.
24. Seizinger BR, Martuza RL, Gusella JF. Loss of genes on chromosome 22 in tumorigenesis of human acoustic neuroma. Nature 1986; 322: 644–47.
25. Gutmann DH. Molecular insights into neurofibromatosis 2. Neurobiol Dis 1997; 3: 247–61.
26. Sheikh HA, Tometsko M, Niehouse L, et al. Molecular genotyping of medullary thyroid carcinoma can predict tumor recurrence. Am J Surg Pathol 2004; 28: 101–6.
27. Claudio JO, Lutchman M, Rouleau GA. Widespread but cell type-specific expression of the mouse neurofibromatosis type 2 gene. Neuroreport 1995; 6: 1942–46.
28. Giovannini M, Robanus-Maandag E, Van der Valk M, et al. Conditional biaalelic NF2 mutation in the mouse promotes manifestations of human neurofibromatosis type 2. Genes Dev 2000; 14: 1617–30.
29. McClatchey AI, Saotome I, Mercer K, et al. Mice heterozygous for a mutation at the NF2 tumor suppressor locus develop a range of highly metastatic tumors. Genes Dev 1998; 12: 1121–33.
30. Tikoo A, Varga M, Ramesh V, et al. An anti-Ras function of neurofibromatosis type 2 gene product (NF2/merlin). J Biol Chem 1994; 269: 23387–90.
31. Lutchman M, Rouleau GA. The neurofibromatosis type 2 gene product, schwannomin, suppresses growth of NIH 3T3 cells. Cancer Res 1995; 55:2270–74.
32. Gutmann DH, Geist RT, Xu H, et al. Defects in neurofibromatosis 2 protein function can arise at multiple levels. Hum Mol Genet 1998; 7: 335–45.
33. Gutmann DH, Sherman L, Seftor L, et al. Increased expression of the NF2 tumor suppressor gene product, merlin, impairs cell motility, adhesion and spreading. Hum Mol Genet 1999; 8: 267–75.
34. Gusella JF; Ramesh V, MacCollin M, et al. Merlin: the neurofibromatosis 2 tumor suppressor. Biochem Biophys Acta 1999; 1423: M29-M36.
35. Ramesh V. Merlin and the ERM proteins in Schwann ceils, neurons and growth cones. Nat Rev Neurosa 2004; 5: 462–70.
36. Vaheri A, Carpen O, Heiska L, et al. The ezrin protein family: membrane-cytoskeleton interactions and disease associations. Curr Opin Cell Biol 1997; 9: 659–66.
37. Xiao GH, Beeser A, Chernoff J, et al. p21-activated kinase links Rac/Cdc2 signaling to merlin. J Biol Chem 2002; 277: 883–86.
38. Rong R, Tang X, Gutmann DH, et al. Neurofibromatosis 2 (NF2) tumor suppressor merlin inhibits phosphatidylinositol 3-kinase through binding to PIKE-L. Proc Natl Acad Sci U S A 2004; 101: 18200–205.
39. Muranen T, Gronholm M, Renkema GH, et al. Cell cycle-dependent nucleocytoplasmic shuttling of the neurofibromatosis 2 tumor suppressor merlin. Oncogene 2005; 24: 1150–58.
40. McClatchey AI, Giovannini M. Membrane organization and tumorigenesis – the NF2 tumor suppressor, Merlin. Genes Dev 2005; 19: 2265–77.
41. Weinberg RA. Tumor suppressor genes. Science 1991; 254: 1138–46.
42. Zheng l, Lee WH. The retinoblastoma gene: a prototypic and multifunctional tumor suppressor. Exp Cell Res 2001; 264: 2–18.
43. Jacks T, Fazeli A, Schmitt EM, et al. Effects of an Rb mutation in the mouse. Nature 1992; 359: 295–300.
44. Lee EY, Chang CY, Hu N, et al. Mice deficient for pRb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 1992; 359: 288–95.
45. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70.
46. Friend SH, Bernards R, Rogelj S, et al. A human cDNA segment with properties of the gene that predisposes to retinoblastoma and osteosarcoma. Nature 1986; 323: 643–46.
47. Lee EY, To H, Shew JY, et al. Inactivation of the retinoblastoma susceptibility gene in human breast cancers. Science 1988; 241: 218–21.
48. Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene 1999; 18: 7873–82.
49. Lee WH, Bookstein R, Hong F, et al. Human retinoblastoma susceptibility gene: Cloning, identification, and sequence. Science 1987; 235: 1394–99.
50. Hong FD, Huang HJ, To H, et ab Structure of the human retinoblastoma gene. Proc Natl Acad Sci USA 1989; 86: 5502–06.
51. Kennedy BK, Barbie DA, Classon M, et al. Nuclear organization of DNA replication in primary mammalian cells. Genes Dev 2000; 14: 2855–68.
52. Wang JY. Retinoblastoma protein in growth suppression and death protection. Curr Opin Genet Dev 1997; 7: 39–45.
53. Classon M, Dyson N. pl07 and pl30: versatile proteins with interesting pockets. Exp Cell Res 2001; 264: 135–47.
54. Ichimura K, Hanafusa H, Takimoto H, et al. Structure of the human retinoblastoma-related p107 gene and its intragenic deletion in a B-cell lymphoma cell line. Gene 2000; 251: 37–43.
55. Claudio PP, Howard CM, Pacilio C, et al. Mutations in the retinoblastoma-related gene RB2/p130 in lung tumors and suppression of tumor growth in vivo by retrovirus-mediated gene transfer. Cancer Res 2000; 60: 372–82.
56. Cinti C, Leoncini L, Nyongo A, et al. Genetic alterations of the retinoblastoma-related gene RB2/pl30 identify different pathogenic mechanisms in and among Burkitt's lymphoma subtypes Am J Pathol 2000; 156: 751–60.
57. Harrington EA, Bruce JL, Harlow E, et al. pRb plays an essential role in cell cycle arrest induced by DNA damage. Proc Natl Acad Sci USA 1998; 95: 11945–50.
58. Ludlow JW, DeCaprio JA, Huang C-M, etal. SV40 large T antigen binds preferentially to an underphosphoryiated member of theretinoblastoma susceptibility gene product family. Cell 1989; 56: 57–65.
59. DeCaprio JA, Ludlow JW, Lunch D, et al. The product of the retinoblastoma susceptibity gene has properties of a cell cycle regulatory element. Cell 1989; 58: 1085–95.
60. Chen PL, Scully R Shew JY, et al. Phosphorylation of the retinoblastoma gene product is modulated during the cell cycle andcellular differentiation. Cell 1989; 58: 1193–98.
61. Buchkovich K, Duffy LA, Harlow E. The retinoblastoma protein is phosphorylated during specific phases of the cell cycle. Cell 1989; 58: 1097–1105.
62. Lee JO, Russo AA, Pavletich NP. Structure of the retinoblastoma tumor-suppressor pocket domain bound to a peptide from HPV E7. Nature 1998; 391: 859–65.
63. Dyson N. pRb, pl07 and the regulation of the E2F transcription factor. J Cell Sci Suppl 1994; 18:81–7.
64. Morris EJ, Dyson NJ. Retinoblastoma protein partners. Adv Cancer Res 2001; 82: 1–54.
65. Weinberg RA. The retinoblastoma protein and cell cycle control. Cell 1995; 81: 323–30.
66. Goodrich DW, Wang NP. Qian YW, et al. The retinoblastoma gene product regulates progression through the G1 phase of the cell cycle. Cell 1991; 67: 293–302.
67. Hinds PW, Mittnacht S, Dulie V, et al. Regulation of the retinoblastoma protein functions by ectopic expression of human cyclins. Cell 1992; 70: 993–1006.
68. Sherr CJ. Cancer cell cycles. Science 1996; 274: 1672–77.
69. Lai A, Lee JM, Yang WM, et al. RBP1 recruits both histone deacetylase-dependent and independent repression activities to retinoblastoma family proteins. Mol Cell Biol 1999; 19: 6632–41.
70. Strobeck MW, Knudsen KE, Fribourg AF, et al. BRG1 is required for RB-mediated cell cycle arrest. Proc Natl Acad Sci USA 2000; 97: 7748–53.
71. Sherr CJ. G1 phase progression: cycling on cue. Cell 1994; 79: 551–55.
72. Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol 1998; 18: 753–61.
73. Lees JA, Buchkovich KJ, Marshak DR, et al. The retinoblastoma protein is phosphorylated on multiple sites by human cdc2. EMBO J 1991; 10: 4279–90.
74. Knudsen ES, Wang JY. Differential regulation of retinoblastoma protein function by specific Cdk phosphorylation sites. J Biol Chem 1996; 271: 8313–20.
75. Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13: 1501–12.
76. Ludlow JW, Nelson DA. Control and activity of type-1 serine/threonine protein phosphatase during the cell cycle. Semin Cancer Biol 1995; 6: 195–202.
77. Gope R, Gope ML. Effect of sodium butyrate on the expression of retinoblastoma (RB1) and p53 genes and phosphorylation of retinoblastoma protein in human tumor cell line HT29. Cell Mol Biol 1993; 39: 589–97.
78. Gope ML, Gope R. Effect of sodium butyrate on methyiation pattern of retinoblastoma (RB1) gene in human colon tumor cell line HT29. Curr Sci 2003; 84: 430–34.
79. Slack RS, El-Bizri H, Wong J, et al. A critical temporal requirement for the retinoblastoma protein family during neuronal determination. J Cell Biol 1998; 140: 1497–509.
80. de Bruin A, Wu L, Saavedra HI, et al. Rb function in extraembryonic lineages suppresses apoptosis in the CNS of rb-deficient mice. Proc Natl Acad Sci USA 2003; 100: 6546–51.
81. MacPherson D, Sage J, Crowley D, et al. Conditional mutation of Rb causes cell cycle defects without apoptosis in the central nervous system. Mol Cell Biol 2003; 23: 1044–53.
82. Jiang Z, Guo Z, Saad FA, et al. Retinoblastoma gene promoter directs transgene expression exclusively to the nervous system. J Biol Chem 2001; 276: 593–600.
83. Marino S, Vooijs M, van Der Gulden H, et al. Induction of medulloblastomas in p53-null mutant mice by somatic inactivation of Rb in the external granular layer cells of the cerebellum. Genes Dev 2000; 14: 994–1004.
84. Nowak MA, Komarova NL, Sengupta A, et al. The role of chromosomal instability in tumor initiation. Proc Natl Acad Sci USA 2002; 99: 16226–31.
85. Zheng L, Chen Y, Riley DJ, et al. Retinoblastoma protein enhances the fidelity of chromosome segregation mediated by hsHeclp. Mol Cell Biol 2000; 20: 3529–37.
86. Di Leonardo A, Khan SH, Linke SR et al. DNA rereplication in the presence of mitotic spindle inhibitors in human and mouse fibroblasts lacking either p53 or pRb function. Cancer Res 1997; 57: 1013–19.
87. Yamamoto H, Soh JW, Monden T, et al. Paradoxical increase in retinoblastoma protein in colorectal carcinomas may protect cells from apoptosis. Clin Cancer Res 1999; 5: 1805–15.
88. Gope ML, Chun M, Gope R. Comparative study of the expression of Rb and p53 genes in human colorectal cancers, colon carcinoma cell lines and synchronized human fibroblasts. Mol Cell Biochem 1991; 107: 55–63.
89. Gope R, Gope ML. Abundance and state of phosphorylation of the retinoblastoma susceptibility gene product in human colon cancer. Mol Cell Biochem 1992; 110:123–33.
90. Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev 1998; 12: 2245–62.
91. Sharpless NE, Bardeesy N, Lee KH, et al. Loss of pl6Ink4a with retention of pl9Arf predisposes mice to tumorigenesis. Nature 2001; 413: 86–91.
92. Ma D, Zhou , Harbour JW. Distinct mechanisms for regulating the tumor suppressor and antiapoptotic functions of Rb. J Biol Chem 2003; 278: 19358–66.
93. Jacks T, Weinberg RA. The expanding role of cell cycle regulators. Science 1998; 280: 1035–6.
94. Simpson DJ, Hibberts NA, McNicol AM, et al. Loss of pRb expression in pituatory adenomas is associated with methyiation of the RB1 CpG island. Cancer Res 2000; 60: 1211–16.
95. Nakamura M,Yonekawa Y, Kleihues P, et al Promoter hypermethylation of the RB1 gene in glioblastomas. Lab Invest 2001; 81: 77–82.
96. Mawrin C, Kirches E, Boltze C, et al. Immunohistochemical and molecular analysis of p53, RB, and PTEN in malignant peripheral nerve sheath tumors. Virchows Arch 2002; 440: 610–15.
97. Lasak JM, Welling DB, Akhmametyeva EM, et al. Retinoblastoma-cyclin-dependent kinase pathway deregulation in vestibular schwannomas. Laryngoscope 2002; 112: 1555–61.
98. Xiao GH, Gallagher R, Shetler J, et al. The NF2 tumor suppressor gene product, merlin, inhibits cell proliferation and cell cycle progression by repressing cyclin D1 expression. Mol Cell Biol 2005; 25: 2384–94.
99. Thomas R, Prabhu PD, Mathivanan J, et al. Altered structure and expression of RB1 gene and increased phosphorylation of pRb in human vestibular schwannomas. Mol Cell Biochem 2005; 271: 113–21.
100. Dayalan AHPP, Jothi M, Keshava R, et al. Age dependent phosphorylation and deregulation of p53 in human vestibular schwannomas. Mol Carcinogen 2006; 45: 38–46.
(c) Annals of Neurosciences.All Rights Reserved