Annals of Neurosciences, Vol 14, No 3 (2007)
Annals of Neurosciences, Volume 14, Issue 3 (July), 2007
POSSIBLE ROLE OF THE TUMOR SUPPRESSOR GENE RETINOBLASTOMA (RBI) IN HUMAN BRAIN TUMOR DEVELOPMENT
Corresponding address:
Dr. Rajalakshmi Gope
Additional Professor
Department of Human Genetics
NIMHANS, Bangalore 560 029.
Email:
Abstract
Human brain tumors are a group of tumors that primarily originate from the central nervous system. These tumors vary in their etiology, histology, biology, prognosis and treatment. The overall incident rate of brain tumors are reported to be lower compared to other human tumor types. However, it results in a disproportionately high mortality rate due to cancers. The human retinoblastoma gene (RBI) is the first human tumor suppressor (TS) gene to be identified and it is one of the most widely studied genes in a variety of human cancers. Data from these studies showed that it is one of the most important cell cycle regulatory genes. Inactivation of RBI gene has been reported in various human tumors including brain tumors. Data from the Western population indicates that the TS function of this gene is lost due to deletions, mutations or absence of its protein product in a considerable number of human brain tumor cases. To the contrary, data from the brain tumor tissues of Indian population indicates that the inactivation of RBI gene pathway could occur though a different mechanism. This could abo explain the rarity of retinoblastomas, one of the childhood tumors, in Indian population. In this review we have discussed the possible role RBI gene in the development of human brain tumors.
Key words: Brain tumors, Cellcycle, pRb, RBI, Tumor suppressor gene
Introduction
Human brain tumors account for only 2% of all human cancers but they have a disproportionately high mortality rate compared to the other tumors. The human brain tumors are difficult to manage and treat. The overall survival of brain tumor patients is less than two years from the time of diagnosis. Inspite of advancement in diagnostic and treatment procedures the survival rate has not changed over the past four decades. It is now clear that most cancers arise due to a complex interplay between various oncogenes and tumor suppressor genes. The human retinoblastoma gene (RBI) is an important tumor suppressor gene. The available literature indicates the importance on RBI gene in neuronal development and differentiation. In this review we have evaluated possible role of RBI gene in human brain development and provide current status in these subjects.
Human Brain Tumors
Human brain tumors are a collection of intracranial tumors formed due to abnormal or uncontrolled cell division where each tumor type has its own etiology, biology, prognosis and treatment (1). Brain tumors commonly arise from neurons, glial cells (astrocytes, oligodendrocytes and ependymal cells), lymphatic tissue, blood vessels, cranial nerves (myelin-producing Schwann cells), meninges, skull, pituitary and pineal gland (2).
Classification of brain tumors
The current WHO (World Health Organisation) classification of brain tumor consists of more than 120 intra cranial tumors classified under 7 major categories which includes tumors of neuroepithelial tissue, peripheral nerves, meninges, lymphomas and hemopoietic neoplasms, germ cell tumors, tumors of the sellar region and metastatic tumors (3,4).
Primary intra cranial tumors
Primary human intra cranial tumors are true brain tumors, arising exclusively from cells normally present in the brain. Gliomas originate from glial cells like astrocytes (astrocytomas), oligodendrocytes (oligodendrogliomas), or ependymal cells (ependymoma). Some tumors consist of cells of astrocytic and oligodendroglial origin and these are known as mixed gliomas or oligoastrocytomas. Additional ly, (a) mixed glio-neuronal tumors, tumors displaying a neuronal, as well as a glial component, e.g. gangliogliomas, disembryoplastic neuroepithelial tumors and (b) tumors originating from neuronal cells e.g. gangliocytoma, central gangliocytoma also exist (5).
From the histological perspective, astrocytomas, oligondedrogli-omas, and oligoastrocytomas may be benign or malignant. Glioblastoma multiforme (GBMs) represents the most aggressive form of malignant glioma (2,6). Other primary brain tumors include primitive neuroectodermal tumors (PNETs) such as medulloblastoma, medulloepithelioma, neuroblastoma, retinoblastoma and ependymoblastoma, tumors of the pineal parenchyma (e.g. pineocytoma, pineoblastoma), ependymal cell tumors, choroid plexus tumors, neuroepithelial tumors of uncertain origin (e.g. gliomatosis cerebri, astroblastoma) etc.
Another type of primary intra cranial tumor is primary cerebral lymphoma, also known as primary CNS lymphoma, which is a type of non-Hodgkin's lymphoma that is much more prevalent in those with severe immuno-suppression disorder such as AIDS.
Secondary intra cranial tumors
Secondary brain tumors originate from malignant tumors located primarily in other organs. Their incidence is higher than that of primary brain tumors (2). The most frequent types of metastatic brain tumors originate from primary tumors of lung, breast, skin, kidney, and colon and these are known to occur through the bloodstream (7).
Incidence of human brain tumors
Gliomas represent the largest proportion of primary brain tumors, accounting for approximately 50% of all brain tumors. Meningiomas are the second most common brain tumors comprising approximately 20 to 25%, followed by pituitary adenomas, nerve sheath tumors and primary CNS lymphoma each of which represent less than 10% of all primary brain tumors (2,5,8).
The overall incidence rate for primary brain and central nervous system tumors is reported to be 14.8 for every 100,000 individuals each year. The overall incidence rate is 4.3 in 100,000 each year for children new born to 19 years of age, 4.4 for 100,000 population each year for children less than 15 years and 19.0 for 100,000 each year for adults 20 years or older (9).
Age and gender distribution
The overall incidence of all primary malignant brain tumors combined is higher among males, which are known to be 8.7 for 100,000 population each year, compared to females which is 6.2 for every 100,000 population each year. The annual incidence of all primary non-malignant brain tumors combined is higher among females, that is, 8.8 for every 100,000 population each year compared to males, which is, 5.8 for 100,000 population each year (9).
Distribution of brain tumors shows that the incidence is highest among children. In addition, the tumor incidence is known to increase exponentially from early twenties until the age of 70 years then is reported to decline with increasing age (2).
Survival
Brain tumors accounts for approximately 2% of all human cancers but it is the fifth leading cause of death due to cancer. The five-year relative survival rate following diagnosis of a primary malignant brain tumor, including lymphoma, tumors of the pituitary and pineal gland, and olfactory tumors of the nasal cavity is 28.1% for males and 30.5% for females. The five-year relative survival rates following diagnosis of a primary malignant brain tumor for patients of 9 years and below is reported to be 64.8%, age 20 to 44 years is 47,9%, age 45 to 64 years is 23.1%, age 65 to 74 years is 6.6% and 75 and above is 4.8%. The estimated five and ten-year relative survival rates for malignant brain tumors are 28% and 24% respectively. (9,10).
Causes
Apart from vinyl chloride exposure or ionizing radiation, there are no known environmental factors associated with brain tumors. Irradiation of the cranium even at the low doses can increase the incidence of meningiomas by a factor of 10 and the incidence of glial tumors by a factor of 3 to 7 with the latency period of 10 years to 20 years after exposure (1). Mutations and deletions of tumor suppressor genes are documented in many human brain tumor types. Patients with various inherited diseases, such as Von Hippel-Lindau syndrome, multiple endocrine neoplasia, and neurofibromatosis type 2 (NF2) are at higher risk of developing brain tumors (11). Recently, use of cellular phones and hair dye, living close to high tension wires, head trauma and dietry exposure to N-nitrosourea or other nutritional factors have been reported to increase the risk of brain tumors however, these results are not conclusive (12–14).
Symptoms
Symptoms associated with brain tumors depend on the type of tumor, tumor size and location. Onset of symptom during the tumor progression correlates in many cases with the tumor type. Slow-growing benign tumors have late symptom onset and fast-growing malignant tumors have early onset of symptoms. Many low-grade tumors can remain asymptomatic for several years. Onset of epilepsy is a frequent reason for seeking medical attention in brain tumor cases (2,15).
Large tumors or tumors with extensive perifocal swelling and edema inevitably lead to increased intracranial pressure (IICP). IICP translates clinically into headaches, nausea and vomiting, altered state of consciousness such as somnolence and coma. However, even small tumors obstructing the passage of cerebrospinal fluid (CSF) may cause early signs of increased intracranial pressure (2,15).
Depending on the tumor location and the damage it has caused any type of focal neurologic symptoms such as cognitive and behavioral impairment, personality changes, hemiparesis, hypesthesia, aphasia, ataxia, visual field impairment, facial paralysis, double vision, tremor etc may occur. These symptoms are however not specific for brain tumors as they may also be caused by a variety of neurologic conditions such as stroke and traumatic brain injury. However, location of the tumors generally affects the normal functioning of motor, sensory and visual systems (15). A bilateral temporal visual field defect, bitemporal hemianopia arise due to compression of the optic chiasm. This often is associated with the endocrine dysfunction like hypo or hyperpituitarism and hyperprolactinemia due to pituitaiy tumor (2).
Diagnosis
Imaging plays a central role in the diagnosis of brain tumors. Non-invasive, high-resolution modalities, such as computed tomography (CT) (Figure 1) and magnetic resonance imaging (MRI) are used frequently to diagnose brain tumors (16). Electrophysiological procedures, such as electroencephalography (EEG) play a marginal role in the diagnosis of brain tumors. Histological examination is essential for determining the appropriate diagnosis, treatment and prognosis.
Histopathology
Diffused astrocytomas show mild atypia particularly nuclear pleomorphism and hyper chromassia (17). The cells of fibrillary astrocytomas may appear as bare nuclei and they show astrocyticdifferentiation and exhibit prominent fibrillary stretch of eosinophilic cytoplasm or a gemistocytic appearance (2,3).
Cytological and nuclear pleomorphism, increased nuclear: cytoplasm ratio, multi lobules of nuclei and greater range of cell sizes may be apparent in the anaplastic astrocytomas. Enhanced mitotic activity and neovascularisation distinguish the anaplastic astrocytomas from other astrocytomas. No necrosis is found in anaplastic astrocytomas (Figure 2). Necrosis and a florid microvascular proliferation is the key feature of GBMs. Cellular pleomorphism is more extreme in GBMs than the anaplastic astrocytomas. GBMs often show increased cellularity surrounding a region of necrosis called a 'pseudopalisading'. Ischaemic necrosis also had been found in GBMs (3).
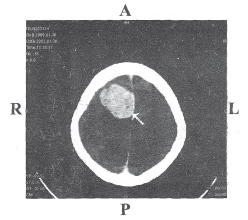
Figure 1: CT image of a 40 year old female with meningothelial meningioma grade III. The arrow indicates the tumor of size 18/13 mm at the temporal lobe. R- Right; L- Left; A- Anterior; P- Posterior
Image courtesy: Department of Neuroimaging and Interventional Radiology, NIMHANS
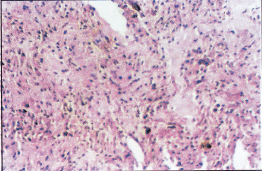
Figure 2: Hematoxylin and Eosin stained section of an anaplastic astrocytoma grade III showing nuclear pleomorphism and increased cellularity.
Oligodendrogliomas are best recognized by the classic honeycomb appearance of uniform collections of cells with round nuclei and a distinctive perinuclear halo that occurs as an artifact of tissue handling. These tumor cells occasionally adopt slightly elongated structure but the nucleus retains the characteristics uniformly (3). Gangliogliomas consist of disorganized ganglion cells set against a neuropil-like background on a lacy fibrillary background containing a few reactive astrocytes.
Meningioma exhibits a wide rage of histologic pattern (Figure 3) furthermore a single meningioma can also show a combination of hitological pattern. Menigothelial meningioma consists of sheets of lobule of oval cells that form rudimentary menigothelial whorls in a few places. Fibroblastic meningiomas consist of spindle shaped cells associated with variable amount of perinuclear collagen. Menigothelial and fibrous areas are combined in transitional meningioma, which contain widespread whorls and scattered psammoma bodies (3).
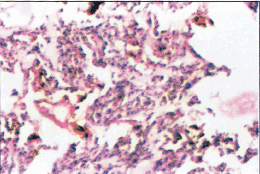
Figure 3: Hematoxylin and Eosin stained section of meningioma grade III showing nuclear pleomorphism, numerous mitoses and necrosis.
Molecular genetic alterations in brain tumors
In general, cancers develop due to series of discrete genetic and molecular alterations that lead to stepwise accumulation of abnormalities in the fundamental cell regulatory pathways (18). Many of these defects act on growth factor signaling pathways that tightly regulate various cellular functions such as cell division, cell survival, differentiation, cell-cell and cell-matrix interactions and angiogenesis. Under normal circumstances these growth factors control tissue growth, differentiation and repair in response to injury. Deregulation of these pathways by either direct or indirect involvement of proto-oncogenes and/or tumor suppressor genes result in oncogenic transformation, uncontrolled cell proliferation and subsequently cancer.
Mutational activation or overexpression of proto-oncogenes is now recognized to play a fundamental role in initiation and progression of many human brain tumor types (18–20). Conversely, tumor suppressor (TS) genes code for proteins that down regulate oncogenic pathway under normal circumstances. Loss of function or inactivation of tumor suppressor genes isequally important to proto-oncogene activation during tumorigenesis. The interplay between the TS genes and proto-oncogenes during cell cycle and differentiation is a highly complex process (20).
Overexpression of cyclin dependent kinases (cdks) and loss of expression of cyclin-dependent kinase inhibitors play important role during malignant progression of many human tumors including the gliomas (19). There is increasing evidence that inactivation of cell cycle regulatory genes is a frequent event in human astrocytic brain tumors. Homozygous deletion of pl6 (CDKN2/MTS I) gene has been reported in 30 to 60% of glioblastomas. Cyclin dependent kinase 4 (CDK4) amplification is identified in 11 to 15% of glioblastomas while loss of retinoblastoma gene (RB1) locus (LOH) is reported in many glioblastomas. Loss of pRb expression is detected by western blot in 22% of malignant gliomas and by immunohistochemistry in one third of high-grade astrocytomas with LOH on chromosome 13q. Furthermore, RB1 gene mutations have been found in 12 to 20% glioblastomas (21).
Loss of p53 function often occurs early in the course of glioma development (22–24). Possibly the most frequently mutated tumor. suppressor gene in human cancers, p53 normally plays a fundamental role in preventing DNA injury and subsequent oncogenesis through two major pathways: (a) inhibition of the cell cycle and (b) induction of apoptosis (25). Expression of pl4ARF is regulated by the E2F1 transcription factor that is activated under conditions of uncontrolled cell proliferation. pl4ARF reverses Mdm2-mediated p53 inactivation and inhibits Mdm2-mediated p53 degradation (26,27). Thus, pl4ARF limits uncontrolled cell cycle progression even in presence of Mdm2 expression. Genetic mutations leading to excessive Mdm2 activity and loss of pl4ARF functions are reported to be common in human brain tumors (28).
Treatment and prognosis
Treatment involves combination of surgery, radiationtherapy and chemotherapy. Multiple metastatic tumors are generally treated with radiationtherapy and chemotherapy. Survival rates in primary brain tumors depend on the type of tumor, age, functional status of the patient, extent of surgical tumor removal, just to mention a few. Patients with benign gliomas may survive for many years while survival in most cases of GBM, that is, the most malignant form, is limited to a few months after diagnosis (2). In more difficult cases, stereotactic radiotherapy (gamma knife) remains a viable option. Assortments of other treatments are commonly used when a brain tumor fails to respond to surgery, radiationtherapy or chemotherapy. These involve the use of angiogenesis inhibitors, differentiating agents, immunotherapy, and gene therapy (2).
Recent advances in nanotechnology are being used for developing new, efficient magnetic nano-vectors for brain tumor therapy (29,30). There are reports of rats with cancer that received traditional Photofrin therapy surviving 13 days, while rats treated with the Photofrin/nanoparticle method surviving longer, that is, average of 33 days. In this study 40 percent of the rats remained disease-free six months after treatment (30). Thus, use of small biological particles such as Photofrin/nanoparticles could provide an opportunity to successfully diagnose and treat brain tumors and this could also preserve healthy brain tissues and more effectively destroy tumor cells only.
Human Retinoblastoma Gene (RBI)
Discovery of tumor suppresser (TS) genes
Presence of tumor suppresser (TS) genes was identified using cell fusion studies. In 1957 Harris and coworkers fused tumor cells with normal fibroblast, lymphocyte and keratinocytes and found that the genetic factor present in the normal cells suppressed malignant property of the tumor cells (31). In 1987 Weissman et al introduced a single chromosome into the tumor cells by microcell fusion-mediated transfer, which also inhibited the tumorigenecity (32). Results from these experiments lead to the discovery of existence of tumor suppresser genes.
In 1971 Knudson proposed that sporadic retinoblastomas could result from as few as two mutational events occurring in both alleles of the same gene and in familial retinoblastomas one defective allele is inherited from the parent and the second 'hit' could occur at the somatic level (33). Genetic analysis of retinoblastomas revealed that the chromosome region 13ql4 is frequently lost in both hereditary and sporadic form of these tumors (33). Later many groups independently isolated, cloned and characterized the RBI gene from this locus. Detailed studies on the RBI gene identified it as a candidate for the retinoblastoma susceptibility and these studies marked the beginning of new era of cancer research (34,35).
Structure of RB1 gene
RB1 gene spans approximately 200 kb and is localized on human chromosome 13ql4. It has 27 exons ranged in length from 31 (exon 24) to 1889 nucleotides (exon 27) (34–36). The sizes of the introns range from 80 base pair (intron 15) to approximately 17 kb (intron 17). Exon 1 contains first methionine and 5' untranslated sequences. The open reading frame stops 140 bases from the 5' end. Two potential initiation codons exist in the open reading frame, one at 139 and another at 145. The promoter region lies between +13 to +83 (34) and it contains no TATA or CCAAT box motifs. The 5' region of RB1 gene is rich in its GC content (34).
The retinoblastoma protein (pRb)
The RB1 gene encodes a relatively stable nucleophosphoprotein, pRb, which has 928 amino acids and a molecular weight of 110 kDa (37). pRb has a half life of more than 8 hours (38). The level of pRb is relatively constant however its function is modulated by posttranslational modifications of the 16 potential phosphorylation sites (39–41).
Localization of pRb within the nucleus
High-resolution deconvolution microscopic studies have revealed that during G1 and S phases, the three RB family of proteins(RB1, RB2/p130 and p107) are found in the perinucleolar foci (42). pRb is known to be associated with the nuclear matrix only during G0 and G1 phases (43).
RB pocket family proteins
Similar to RB1, the RB2/p130 and p107 are members of RB family. Together these are termed as pRb family, also known as pocket family protein because of their structural similarities, particularly at the A and B domains. p107 and p130 are closely related to each other and have 47% identity whereas 21% identity with RB1 at the amino acid level. The similarities between them are more at the A and B domains i.e. small pocket domain with 30 to 40% homology. The N and C domains are not conserved. The A and the B domains are necessary for the functions of family of pocket proteins (44).
Function pRb protein during cell cycle
Retinoblastoma protein (pRb) acts as a central regulatory component of the cell cycle machinery and it regulates cell division at the G1/S checkpoint (45). pRb acts as a check point, that is controlled during the transition of cell cycle from early G1 phase to late G1/S phase (46,47). Whenever pRb gets hyperphosphorylated it becomes inactive and releases E2F free to activate its target genes. The E2F family of proteins has the ability to bind to the promoter region of many genes that are necessary for cell cycle progression and they are repressed by pRb once the cell cycle is completed. Progression of cell cycle, transition of cells from G1 to S phase in particular, requires phosphorylation of pRb and it is carried out by cyclin dependent kinases (CDKs) in association with the corresponding cyclins. pRb becomes active through hypophosphorylation once the cell exits from M phase which is again able to bind to E2F and control cell division at G1/S checkpoint (46,47).
Function of pRb in other phases of cell cycle
Studies showing over expression of pRb in S phase and arrested cells at G2 phase suggest a role for pRb in G2/M progression (48). Rb/E2F mediated G1 phase regulation may also coordinate the mechanism that controls the G2/M transition (49). When cells have entered S phase Rb/E2F complex repress the transcription of cyclin A, which is involved in mitosis. pRb is also known to be involved in mitosis exit (50).
Phosphorylation site-mutated pRb (PSM-Rb) not only has the capability of arresting cells at G1 phase, but also is able to inhibit S phase progression. Such an inhibitory effect cannot be bypassed through overexpression of Gl cyclins such as cyclin E, suggesting a distinct role of pRb in S phase (51). These studies suggest that pRb is involved in the progression of the entire cell cycle.
pRb in transcriptional repression
pRb is known to repress transcription of a set of genes that are necessary for the S phase, thus preventing S phase progression when cells are in the resting phase. Transcriptional repression of pRb is carried out by its interaction with the E2F family of transcription factors. pRb also associates with several proteins involved in transcriptional repression such as histone deacetylases HDAC1, HDAC2, and HDAC3 (52,53).
pRb in genomic stability
Genomic stability in a cell is maintained by accurate DNA replication, segregation of chromosomes and high fidelity of DNA repair (54). Inactivation of pRb leads to multiple genetic alterations which inturn enables the cells to become susceptible to cancer.
pRb has an important role in chromosomal segregation. Therefore, presence of functional RB1 gene is essential for proper segregation of chromosomes during mitosis. pRb is known to interact with two groups of proteins via interactions with mitosin, a structural component of the kinetochore and Hec1, a conserved regulator of multiple mitotic events (55). pRb is also known to physically interact with topoisomerase II, which is involved in DNA replication and chromosome segregation (56).
Role of pRb during cell differentiation
Function of pRb is known to be essential for the terminal differentiation of many tissue types (38). pRb accumulates during embryonic development and cell differentiation. pRb coordinates proliferation and differentiation during terminal differentiation in myogenesis, osteogenic differentiation, and adipogenesis (57). pRb is required for terminal differentiation of cerebellar granule cells, and keratinocytes (58). In vitro studies also have demonstrated a role for pRb in terminal adipocyte differentiation (59).
pRb in neural cell differentiation
pRb deficient mice embryos have abnormal neuronal differentiation as displayed by decreased or absent expression of a group of neuronal differentiation markers such as p75 NGFR, âII-tubulin, and Trk A in the trigeminal ganglia and dorsal root ganglia. Primary cultures of neuronal cells from these regions of Rb deficient mouse embryos show decreased outgrowth of axons compared with similar cultures from Rb wild type mouse embryos. The phenotype in the nervous system of Rb-/- embryos has been almost fully characterized. Extensive ectopic cell cycle entry and elevated apoptosis levels were apparent in both central (CNS) and peripheral nervous system (PNS) of these mice by embryonic day 12, that is E12. During mouse embyo development neuronal precursor cells normally exit from cell cycle and begin to differentiate at the time Rb-/- CNS/PNS phenotypes were initiated which suggested pRb might be required during neurogenesis (60,61).
Role pRb in apoptosis
Rb null mice display various defects such as extensive apoptosis. Wild type pRb acts as a survival factor as is evident from the massive cell death observed in pRb deficient mice in tissues where pRb is normally highly expressed (62). Because pRb is activated on exit of the cell cycle during differentiation, it is thought that the function of pRb is to protect differentiating cells from apoptosis.
Several studies provide evidence that pRb may also regulate apoptosis negatively (63–66). Previous reports from colon tumors, cultured colon tumor cell lines, breast cancer, bladder cancer, NF2 tumors and melanoma indicated tumor specific increase in the level of pRb and the phosphorylated form of pRb (39,40,67–70). A positive role for pRb in malignant transformation is also implied in human colon cancer, breast and bladder cancer (67,68,71), perhaps due its antiapoptotic function.
Regulation of pRb functions by phosphorylation
The function of pRb is regulated by phosphorylation and dephosphorylation of serine/threonine residues. There are sixteen serine/threonine residues in pRb and the phosphorylations of these serine/threonine residues determine the function of pRb during cell cycle (40,41). pRb switches between hypophosphorylated and hyperphosphorylated forms during cell cycle (40,72,73). Cell cycle dependant phosphorylation of pRb in vivo is well establihsed (47,72,73). pRb is shown to be phosphorylated in vitro by cyclin dependant kinases (CDKs) (41,72,73). Response to various growth signals cyclin-cdk complexes phosphorylate pRb during cell cycle.
The antiapoptotic function of pRb seems to oppose its tumor suppressor activity. The anti proliferative and anti apoptotic activities of pRb may represent complementary functions that work in concert to maintain the proliferation rate of cells within limits. As a survival strategy, some cancer cells may exploit this dual role of pRb by phosphorylating sites that regulate tumor suppression but voiding phosphorylation of Ser 567 and consequent apoptotic stimulus (74).
Inactivation of tumor suppressor function of pRb
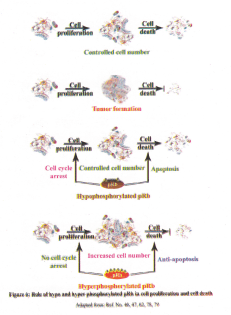
pRb is known to interact with more than 100 protein partners by its multiple protein binding sites and form complexes to perform the normal function (75). Disassembly of these complexes is mediated by four known mechanisms: (a) mutation of the RB1 gene (60); (b) physical binding with viral oncoproteins, such as E1A (76); (c) phosphorylation of pRb (47) and (d) degradation of pRb(77)(Figure 4).
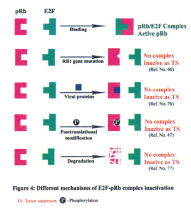
RB1 gene alterations in human tumors
Mutation of the RB1 gene was first observed in inherited retinoblastomas, it is known that loss of Rb function contributes to a wide array of human cancers (47,78). Mutations at the RB1 gene locus lead to absence or altered form of its protein, which has lost its ability to suppress the uncontrolled cell proliferation. Loss of function of pRb eventually leads to tumor development (47). RB1 gene has been implicated in a variety of human tumors like retinoblastomas, oesteosarcomas, small cell lung cancers, breast cancers, bladder cancers etc. (62,78).
Alterations in RB1 gene expression have been reported in many human tumor types including lung cancer, osteosarcoma, leukemia, prostate cancer and bladder cancer (20,46). Increased expression of RB1 mRNA and protein has been reported for many human colon tumor tissues and human colorectal cancer cell lines, breast cancers; brain tumors, VS tumors and bladder cancers (39,40,66,68,71,79–81). Altered phosphorylation of pRb by deregulation of CDK/cyclins and their inhibitors are involved in tumorigenesis.
Alteration of RB1 gene and pRb pathway in brain tumors
The human brain tumors arise from various cell types (Figure 5). RB1 tumor suppressor gene has been studied in several human intracranial tumors including gliomas, meningiomas, pituitary adenomas, vestibular schwannomas etc. There are reports that loss of pRb expression in pituitary tumors and glioblastomas (82) are associated with promoter hyper methylation.
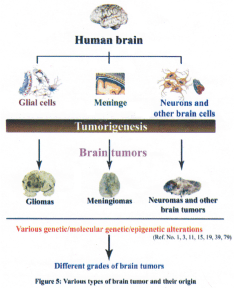
Abnormality in pRb function is reported in 30% of human gliomas (21). Aberrations at the RB1 gene locus were found in high-grade gliomas (83). Allelic loss at the RBI gene and its altered expression has been reported in high-grade astrocytomas (21). Loss of pRb is reported in the progression of pituitary adenoma to carcinoma (84). It was also demonstrated that RB1 gene inactivation and loss of its protein, pRb, might be associated with glial tumor progression (85). LOH of RBI gene was detected in 44% of malignant peripheral nerve sheath tumors (86).
Overexpression of either cyclin D, cdk4/cdk6 in glial tumors may stimulate sustained cdk mediated phosphorylation of pRb (87–89). This may be critical for inactivation of pRb mediated growth suppression and may play an important role in brain tumor progression (67,69,70).
Possible role of RB1 gene in human brain tumors
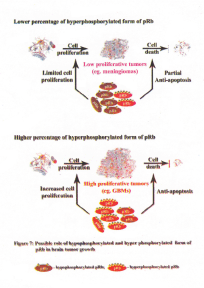
Recent results showed LOH at the RB1 gene locus in high-grade gliomas. In addition, the level of RB1 mRNA was increased in these tumors. Notably, 50% and higher percentage of the total pRb was present as hyperphosphorylated form in almost all the gliomas. Among the meningiomas, approximately 50% of the tumors had increased level of RB1 mRNA and in the remaining tumors the levels were equal to that of the control WI38 cell line. Meningiomas also had increased percentage of hyperphosphorylated pRb compared to the WI38 control cell line. However, in approximately half of the meningiomas studied less than 50% of total pRb was present as hyperhosphorylated form (79) (Figure 6).
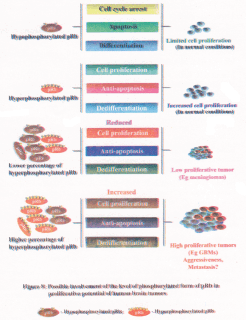
Recent studies showed increased percentage of hyperphosphorylated form of pRb in human brain tumor tissues compared to the control WI38 cell line and it is conceivable that the hyperphosphorylated pRb could control the proliferation of various brain tumor cells to a varying degree (79). The percentage of hyperphosphorylated form of pRb varied among the brain tumor types, which is suggestive of deregulated pRb in these tumor types. It is conceivable that the pRb could play a role in apoptosis in tumors where less than 50% pRb exist in hyperphosphorylated form. Therefore, the variable ratio between the hypophosphorylated form and the hyperphosphorylated form of pRb found in various brain tumor tissues could determine the variable growth potentials (fast or slow) of these tumors (79,90). Gliomas are known to have a higher proliferative potential compared to meningiomas. Presence of higher percentage of hyperphosphorylated form of pRb in almost all gliomas compared to meningiomas could provide an accelerated proliferative potential for these tumors. In addition, presence of increased percentage of hyperphosphorylated form of pRb could also aid in tumor cell survival perhaps due to its anti-apoptotic functions (79) (Figure 7). Thus, the increased percentage of hyperphosphorylated form of pRb found in gliomas could aid in aggressive proliferation of these tumors and could also aid in tumor metastasis (Figure 8). Presence of lower percentage of total pRb as hyperphosphorylated form in meningiomas could induce apoptosis (due to increased percentage of hypophosphorylated pRb), which in turn could lead to slow proliferative potential of meningiomas (Figure 7). Overall, the existence of increased percentage of total pRb as hyperphosphorylated form as compared to the control WI38 cell line is predictive of its role in keeping the tumor cells in dedifferentiated state (91). In addition, it appears that the ratio between the hypophosphorylated form and hyperphosphorylated form of pRb could predict the tumor cell growth, survival, aggressiveness, expansion and possibly metastasis (Figure 8).
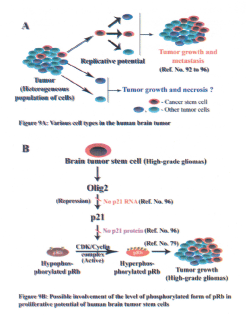
Several studies have shown that many human solid tumors, including brain tumors have small population of cells, which have the characteristics of adult stem cells (92–94). These 'cancer stem cells' have the capacity of self-renewal (Figure 9A). Several reports suggest that these cancer stem cells could be involved in tumorigenic pathways such as initiation, progression and metastasis (92–94).
Presence of cancer stem cells has been reported for brain tumors (95). A recent study by Ligon and coworkers showed that O1ig2, a CNS-restricted-transcription factor is known to regulate the growth of normal CNS stem cells as well as brain tumor stem cells (96). O1ig2 has been shown to directly suppress transcription of p21, which is a kinase inhibitor. p21 is known to inhibit phosphorylation of pRb (78,97,98). It has been shown that gliomas, particularly the malignant gliomas had increased percentage of hyperphosphorylated form of pRb (79) and this could be due to inactivation of p21 by Olig2 in these high-grade tumors (96) (Figure 9B).
Conclusion
The number of brain tumor cases appears to be on the rise in Indian population, which could partially be due to improved screening and diagnostic protocols. It could also be due to increased use of pestiside and change in life style such as change in food habbits and increased consumption of processed food. Inactivation of RBI gene due to gross structural changes that is reported for brain tumors from Western population does not appear to be true for Indian population (79). In addition, it is intriguing that prevalence of retinoblastoma tumors appears to be rare in Indian population compared to that reported for Western population. Data from the brain tumors tissues obtained from Indian population indicate changes such as large deletions, point mutations at the RBI gene locus accompanied by absence of kpRb is not frequent in these tumors. Conversely, RB1 gene deregulation in Indian population appears to be due to changes such as LOH at the intron 1 region, deregulated expression of its mRNA and pRb protein and also due to change in the phosphorylation status of pRb (79). Therefore, we need to develop customized chemotherapeutic protocol as the available drugs and treatment protocols developed for Western population might be less effective or even inappropriate to treat brain tumor patients in India.
Acknowledgement
We thank all the faculty members of the NIMHANS Neurosurgery department for providing the surgical tumor tissues which were used in some of the publications mentioned in this review. Part of the results discussed here are from the research project funded by the Department of Biotechnology (DBT), Government of India, to Dr. R. Gope, project number BT/PRO 703/MED/ 09/133/97. Financial assistance in the form of Junior and Senior Research Fellowships (JRF, SRF) from UGC to MJ and RK are gratefully acknowledged. According to the journal format only limited references are cited in this review. Many more relevant reference could not be cited here and we applogise to the authors whose references are not included.
References
1. DeAngelis LM: Brain tumors. N Engl J Med 2001; 344:114–23.
2. Kaye AH, Laws Jr ER (Eds). In: Brain tumors: An encyclopedic approach. Harcourt publishers Limited, London, 2nd edition, 2001.
3. Kleihues P, Cavanee WK (Eds). WHO classification of tumors, Pathology and genetics: Tumors of the nervous system. Oxford university press, USA, 2nd edition, 2000.
4. Kleihues P, Louis DN, Scheithauer BW, Rorke LB, Reifenberger G, Burger PC, Cavenee WK. The WHO classification of tumors of the nervous system. J Neuropathol Exp Neural 2002; 61: 215–25.
5. Preston-Martin S. Epidemiology of primary CNS neoplasms. Neurol Clin 1996; 14: 273–90.
6. Fisher PG, Buffler PA. Malignant gliomas in 2005: where to GO from here? JAMA 2005; 293: 615–17.
7. Tsukada Y, Fouad A, Pickren JW, Lane WW. Central nervous system metastasis from breast carcinoma. Autopsy study. Cancer 1983; 52: 2349–54.
8. Ohgaki H, Kleihues P. Epidemiology and etiology of gliomas. Acta Neuropathol (Berl) 2005; 109: 93–108.
9. CBTRUS. Statistical Report: Primary Brain Tumors in the United States, 1998–2002. Published by the Central Brain Tumor Registry of the United States, 2006.
10. Ohgaki H, Kleihues P. Population-based studies on incidence, survival rates, and genetic alterations in astrocytic and Oligodendroglial gliomas. J Neuropathol Exp Neurol 2005; 64: 479–89.
11. Bondy M, Wiencke J, Wrensch M, Kyritsis AP. Genetics of primary brain tumors: a review. J Neurooncol 1994; 18: 69–81.
12. Lahkola A, Tokola K, Auvinen A. Meta-analysis of mobile phone use and intracranial tumors. Scand J Work Environ Health 2006; 32: 171–77.
13. Muscat JE, Hinsvark M, Malkin M. Mobile telephones and rates of brain cancer. Neuroepidemiology 2006; 27: 55–6.
14. Nygren C, Adami J, Ye W, et al. Primary brain tumors following traumatic brain injury=Ma population-based cohort study in Sweden. Cancer Causes Control 2001; 12: 733–37.
15. DeAngelis LM, Gutin PH, Leibel SA, Posner JB. Classification, incidence and etiology of intracranial tumors. In: DeAngelis LM, Gutin PH, Leibel SA, Posner JB. Intracranial tumors diagnosis and treatment. Martin Dunitz, London, UK 2002; 3–35.
16. Sundaresan N, Galicich JH. Surgical treatment of single brain metastases from non-small-cell lung cancer. Cancer Invest 1985; 3: 107–13.
17. Prayson RA, Estes ML. Protoplasmic astrocytoma. A clinicopathologic study of 16 tumors. Am J Clin Pathol 1995; 103:705–09.
18. Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet 1993; 9: 138–41.
19. Demuth T, Berens ME. Molecular mechanisms of glioma cell migration and invasion. J Neurooncol 2004; 70:217–28.
20. Hanahan D, Weinberg RA. The hallmarks of cancer. Cell 2000; 100: 57–70.
21. Henson JW, Schnitker BL, Correa KM, et al. The retinoblastoma gene is involved in malignant progression of astrocytomas. Ann Neurol 1994; 36: 714–21.
22. Chen R lavatone A, Fick J, et al. Constitutional p53 mutations associated with brain tumors in young adults. Cancer Genet Cytogenet 1995; 82: 106–15.
23. Okada Y, Hurwitz EE, Esposito JM, et al. Selection pressures of TP53 mutation and microenvironmental location influence epidermal growth factor receptor gene amplification in human glioblastomas. Cancer Res 2003; 63: 413–16.
24. Von Deimling A, Eibl RH, Ohgaki H, et al. p53 mutations are associated with 17p allelic loss in grade II and grade III astrocytoma. Cancer Res 1992; 52: 2987–990.
25. Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307–10.
26. Bates S, Phillips AC, Clark PA, et al. pl4ARF links the tumour suppressors RB and p53. Nature 1998; 395: 124–25.
27. Ichimura K, Bolin MB, Goike HM, et al. Deregulation of the pl4ARF/MDM2/p53 pathway is a prerequisite for human astrocytic gliomas with Gl-S transition control gene abnormalities. Cancer Res 2000; 60: 417–24.
28. Reifenberger G, Liu L, Ichimura K, et al. Amplification and overexpression of the MDM2 gene in a subset of human malignant gliomas without p53 mutations. Cancer Res 1993; 53: 2736–39.
29. Kano MR, Bae Y, Iwata C, et al. Improvement of cancer-targeting therapy, using nanocarriers for intractable solid tumors by inhibition of TGF- signaling. Proc Natl Acad Sci USA 2007; 104: 3460–65.
30. Reddy GR, Bhojani MS, McConville P, et al. Vascular targeted nanoparticles for imaging and treatment of brain tumors. Clin Cancer Res 2006; 12: 6677–86.
31. Harris H, Miller OJ, Klein G, et al. Suppression of malignancy by cell fusion. Nature 1969; 223: 363–68.
32. Weissman BE, Saxon PJ, Pasquale SR, et al. Introduction of a normal human chromosome 11 into Wilms' tumor cell line controls its tumorigenic expression. Science 1987; 236: 175–80.
33. Knudson AG Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc Natl Acad Sci USA 1971; 68: 820–23.
34. Hong FD, Huang HJ, To H, et al. Structure of the human retinoblastoma gene. Proc Natl Acad Sci USA 1989; 86: 5502–06.
35. McGee TL, Yandeil DW, Dryja TP. Structure and partial genomic sequence of the human retinoblastoma susceptibility gene. Gene 1989; 80: 119–28.
36. Pang A, Wu KJ, Hashimoto T, et al. Genomic organization of the human retinoblastoma gene. Oncogene 1989; 4: 401–07.
37. Lee WH, Shew JY, Hong FD, et al. The retinoblastoma susceptibility gene encodes a nuclear phosphoprotein associated with DNA binding activity. Nature 1987; 329: 642–45.
38. Lipinski MM, Jacks T. The retinoblastoma gene family in differentiation and development. Oncogene 1999; 18: 7873–82.
39. Thomas R, Antony Herold Prabhu PD, Mathivanan J, et al. Altered structure and expression of RB1 gene and increased phosphorylation of pRb in human vestibular schwannomas. Mol Cell Biochem 2005; 271 : 113–21.
40. Herold Prabhu, R Thomas, J. Mathivanan, et al. The tumor suppressor gene (RB1) in human vestibular schwannomas. Ann of Neurosci 2006; 13: 113–24.
41. Taya Y. RB kinases and RB-binding proteins: new points of view. Trends Biochem Sci 1997; 22: 14–17.
42. Kennedy BK, Barbie DA, Classon M, et al. Nuclear organization of DNA replication in primary mammalian cells. Genes Dev 2000; 14: 2855–68.
43. Mancini MA, Shan B, Nickerson JA, et al. The retinoblastoma gene product is a cell-cycle dependent, nuclear matrix associated protein. Proc Nati Acad Sci USA 1994; 91: 418–22.
44. Claudio M, Harlow E. The retinoblastoma tumor suppressor in development and cancer. Nat Rev Cancer 2002; 2: 910–17.
45. Wejnberg RA. The retinoblastoma protein and cell cycle control. Cell 1995; 81: 323–30.
46. Jacks T, Weinberg RA. The expanding role of cell cycle regulators. Science 1998; 280: 1035–36.
47. Sherr CJ. Cancer cell cycles. Science 1996; 274: 1672–77.
48. Karantza V, Marco A, Fay D, Sedivy JM. Overproduction of pRb protein after the G1/S boundary causes G2 arrest. Mol Cell Biol 1993; 13: 6640–52.
49. Lukas C, Sorensen CS, Kramer E, et al. Accumulation of cyclin B1 requires E2F and cyclin-A-dependent rearrangement of the anaphase promoting complex. Nature 1999; 401: 815–18.
50. Khan SH, Wahl GM. p53 and pRb prevent rereplication in response to microtubule inhibitors by mediating a reversible G1 arrest. Cancer Res 1998; 58: 396–401.
51. Knudsen ES, Buckmaster C, Chen TT, et al. Inhibition of DNA synthesis by RB. Effects on G1/S transition and S-phase progression. Genes Dev 1998; 12: 2278–92.
52. Brehm A, Miska EA, McCance DJ, et al. Retinoblastoma protein recruits histone deacetylase to repress transcription. Nature 1998; 391: 597–601.
53. Lai A, Lee JM, Yang WM, et al. RBP1 recruits both histone deacetylase-dependent and independent repression activities to retinoblastoma family proteins. Mol Cell Biol 1999; 19: 6632–41.
54. Flores-Rozas H, Kolodner RD. Links between replication, recombination and genome instability in eukaryotes. Trends Biochem Sci 2000; 25: 196–200.
55. Zheng L, Chen Y, Riley DJ, et al. Retinoblastoma protein enhances the fidelity of chromosome segregation mediated by hsHec1p. Mol Cell Biol 2000; 20: 3529–37.
56. Bhat UG, Raychaudhuri P, Beck WT. Functional interaction between human topoisomerase II alpha and retinoblastoma protein. Proc Natl Acad Sci USA 1999; 96: 7859–64.
57. Thomas DM, Carty SA, Piscopo DM, et al. The retinoblastoma protein acts as a transcriptional coactivator required for osteogenic differentiation. Mol Cell 2001; 8: 303–16.
58. Ruiz S, Segrelles C, Bravo A, et al. Abnormal epidermal differentiation and impaired epithelial-mesenchymal tissue interactions in mice lacking the retinoblastoma relatives pl07 and pl30. Development 2003; 130: 2341–53.
59. Richon VM, Lyle RE, McGehee RE Jr. Regulation and expression of retinoblastoma proteins p107 and p130 during 3T3-L1 adipocyte differentiation. J Biol Chem 1997; 272: 10117–24.
60. Jacks T, Fazeli A, Schmitt EM, et al. Effects of an Rb mutation in the mouse. Nature 1992; 359: 295–300.
61. Lee EY, Chang CY, Hu N, et al. Mice deficient for pRb are nonviable and show defects in neurogenesis and haematopoiesis. Nature 1992; 359: 288–95.
62. Classon M, Harlow E. The retinoblastoma tumor suppressor in development and cancer. Nat Rev Cancer 2002; 2: 910–17.
63. Adegbola O, Pasternack GR. Phosphorylated retinoblastoma protein complexes with pp32 and inhibits pp32-mediated apoptosis, J Biol Chem 2005; 280: 15497–502.
64. Haas-Kogan DA, Kogan SC, Levi D, et al. Inhibition of apoptosis by the retinoblastoma gene product. EMBO J 1995; 14: 461–72.
65. Haupt Y, Rowan S, Oren M. p53-mediated apoptosis in HeLa cells can be overcome by excess pRB. Oncogene 1995; 10: 1563–71.
66. Yamamoto H, Soh J-W, Monden T, et al. Paradoxical increase in retinoblastoma protein in colorectal carcinomas may protect cells from apoptosis. Clin Cancer Res 1999; 5: 1805–15.
67. Chatterjee SJ, George B, Goebell PJ, et al. Hyperphosphorylation of pRb: a mechanism for RB tumour suppressor pathway inactivation in bladder cancer. J Pathol 2004; 203: 762–70.
68. Gope R, Gope ML. Abundance and state of phosphorylation of the retinoblastoma susceptibility gene product in human colon cancer. Mol Cell Biochem 1992; 110: 123–33.
69. Liu T, Zhu E, Wang L, et al. Abnormal expression of Rb pathway-related proteins in salivary gland acinic cell carcinoma. Human Pathol 2005; 36: 962–70.
70. Roesch A, Becker B, Meyer S, et al. Overexpression and hyperphosphorylation of retinoblastoma protein in the progression of malignant melanoma. Mod Pathol 2005; 18: 565–72.
71. Botos J, Barhoumi R, Burghardt R, Kochevar DT. Rb localization and phosphorylation kinetics correlate with the cellular phenotype of cultured breast adenocarcinoma cells. In Vitro Cell Dev Biol Anim 2002; 38: 235–41.
72. DeCaprio JA, Furukawa Y, Ajchenbaum F, et al. The retinoblastoma-susceptibility gene product becomes phosphorylated in multiple stages during cell cycle entry and progression. Proc Natl Acad Sci USA 1992; 89: 1795–98.
73. Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol 1998; 18: 753–61.
74. Ma D, Zhou P, Harbour JW. Distinct mechanisms for regulating the tumor suppressor and antiapoptotic functions of Rb. J Biol Chem 2003; 278: 19358–66.
75. Morris EJ, Dyson NJ. Retinoblastoma protein partners. Adv Cancer Res 2001; 82: 1–54.
76. DeCaprio JA, Ludlow JW, Lunch D, et al. The product of the retinoblastoma susceptibity gene has properties of a cell cycle regulatory element. Cell 1989; 58: 1085–95.
77. Diederich L, Fotedar A, Fotedar R. Proteolytic cleavage of retinoblastoma protein upon DNA damage and Fas-mediated apoptosis. Cell Biol Toxicol 1998; 14: 133–40.
78. Bartek J, Bartkova J, Lukas J. The retinoblastoma protein pathway in cell cycle control and cancer. Exp Cell Res 1997; 237: 1–6.
79. Mathivanan J, Rohini K, Gope ML, et al. Altered structure and deregulated expression of the tumor suppressor gene retinoblastoma (RBI) in human brain tumors. Mol Cell Biochem (published online 23rd February 2007).
80. Gope R, Christensen MA, Thorson A et al. Increased expression of the retinoblastoma gene in human colorectal carcinomas relative to normal colonic mucosa. J Natl Cancer Inst 1990; 82: 310–14.
81. Gope ML, Chun M, Gope R: Comparative study of the expression of Rb and p53 genes in human colorectal cancers, colon carcinoma cell lines and synchronized human fibroblasts. Mol Cell Biochem 1991; 107: 55–63.
82. Nakamura M, Konishi N, Hiasa Y, et al. Immunohistochemical detection of CDKN2, retinoblastoma and p53 gene products in primary astrocytic tumors. Int J Oncol 1996; 8: 889–93.
83. Tsuzuki T, Tsunoda S, Sakaki T, et al. Alterations of retinoblastoma, p53, p16 (CDKN2), and p15 genes in human astrocytmas. Cancer 1996; 78: 287–93.
84. Hinton DR, Hahn JA, Weiss MH, Couldwell WT. Loss of Rb expression in an ACTH-secreting pituitary carcinoma. Cancer Lett 1998; 126: 209–14.
85. Hamel W, Westphal M, Shepard HM. Loss in expression of the retinoblastoma gene product in human gliomas is associated with advanced disease. J Neurooncol 1993; 16: 159–65.
86. Mawrin C, Kirches E, Boltze C, et al. Immunohistochemical and molecular analysis of p53, RB, and PTEN in malignant peripheral nerve sheath tumors. Virchows Arch 2002; 440: 610–15.
87. Backlund LM, Nilsson BR, Liu L, et al. Mutations in Rbl pathway-related genes are associated with poor prognosis in anaplastic astrocytomas. Br J Cancer 2005; 93: 124–30.
88. Schmidt EE, Ichimura K, Reifenberger G, Collins VP. CDKN2 (p16/MTS1) Gene Deletion or CDK4 Amplification Occurs in the Majority of Glioblastomas. Cancer Res 1994; 54: 6321–24.
89. Tan PG, Xing Z, Li ZQ. Expression of cyclin D1 in brain gliomas and its significance. Ai Zheng 2004; 23: 63–65.
90. Chau BN, Pan CW, Wang JYJ. Separation of anti-proliferation and anti-apoptotic functions of retinoblastoma protein through targeted mutations of it's A/B domains. PLOS ONE 2006; 1: 82.
91. Gope R, Gope ML. Effect of sodium butyrate on the expression of retinoblastoma (RB1) and p53 gene and phosphorylation of retinoblastoma protein in human colon tumor cell line HT29. Cell Mol Biol 1993; 39: 589–97.
92. Al-Hajj M, Wicha MS, Benito-Hernandez A, et al. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100: 3983–88.
93. Miyamoto T, Weissman IL, Akashi K. AML1/ETO-expressing nonleukemic stem cells in acute myelogenous leukemia with 8; 21 chromosomal translocation. Proc Natl Acad Sci USA 2000; 97: 7521–26.
94. Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature 2001; 414: 105–11.
95. Garraway LA, Sellers WR. Lineage dependency and lineage-survival oncogenes in human cancer. Nat Rev Cancer 2006; 6: 593–602.
96. Ligon KL, Huillard E, Mehta S et al. O1ig2-regulated lineage-restricted pathway controls replication competence in neural stem cells and malignant glioma. Neuron 2007; 53: 503–17.
97. Bansal R, Marin-Husstege M, Bryant M, Casaccia-Bonnefil P. S-phase entry of oligodendrocyte lineage cells is associated with increased levels of p21Cip1. J Neurosa Res 2005; 80: 360–68.
98. Boyian JF, Sharp DM, Leffet L, et al. Analysis of site-specific phosphorylation of the retinoblastoma protein during cell cycle progression. Exp Cell Res 1999; 248: 110–14.
(c) Annals of Neurosciences.All Rights Reserved