Annals of Neurosciences, Vol 17, No 2 (2010)
Annals of Neurosciences, Volume 17, Issue 2 (April), 2010
Juvenile myoclonic epilepsy: EFHC1 at the cross-roads ?
Epilepsy is a complex disease, accounting for up to 1% in the population, and is characterized by recurrent seizures.1, 2 The primary cause for epilepsy could be either genetic or environmental. The former group comprises both polygenic forms and monogenic forms. The heritable Mendelian monogenic forms are, however, rarer traits.1,2 A majority of the genetic forms of epilepsy – both Mendelian and non-Mendelian forms, are classified as "idiopathic epilepsy", a group characterized by the absence of detectable form for brain lesions.1,2 The primary symptom of epilepsy, the epileptic seizure, could either be provoked by specific sensory stimulus (for example flash, stress or sound) or could be unprovoked and the phenotype could range from subtle changes in the behaviour (absence seizure) to violent jerks (myoclonic seizures).1-3 Since all form of seizures result from abnormal electrical discharges in the neurons, and since neuronal electrical output is regulated by ion channels, defects in genes coding for ion channel proteins have long been thought to be the genetic cause for the idiopathic forms of epilepsy4. Therefore, epilepsy syndromes are often grouped under "channelo-pathies".4,5 Consistent with this view, there has been an upsurge, in the last one decade, in the discovery of epilepsy-causing mutations in genes encoding for ion channel proteins.6 While a considerable number of genes that code for channel proteins have been identified to be defective in epileptic patients, the number of independent families that carried these mutations were very few and hence such forms are considered as rarer epilepsies.2,7 One of the exceptions to the school of "channelopathy" is the discovery of mutations in the EFHC1 gene in a significant number of families with juvenile myoclonic epilepsy (JME).8-11 JME is one of the most common epilepsy syndromes accounting for 7% of all cases of epilepsy.2,12 The seizures in JME may begin between late childhood and early adulthood, and is characterized by myoclonic seizures usually in the early morning, soon after awakening.2,12 JME patients may also exhibit tonic-clonic seizures and absence seizures.2,12
The JME gene EFHC1 is ubiquitously expressed and codes for an EF-hand-containing calcium binding protein named as myoclonin.8,13 Two splice variants for the EFHC1 gene, resulting from differential splicing of exon 4, have been identified.8 The protein isoforms coded by these variants differ in their carboxyl terminal; the protein coded by the minor isoform is expected to lack the EF-hand although the functional significance of this transcript is not known.8 Intriguingly, JME associated mutations have been identified for both the transcripts.8,11 While missense mutations have been identified for the protein coded by the major transcript, a deletion/frameshift mutation was reported for the minor transcript. JME is thought to manifest as autosomal dominant disorder,8 and consistent with this view all the mutations that are reported so far in EFHC1 were found to show heterozygosity in the affected individuals.8 However whether or not the disease phenotype results from a gain or loss of function of the mutant protein is yet to be clearly established. Moreover, whether or not proteins coded by both the splice variants are involved in the etiology is yet to be tested. Nonetheless, functional studies on the EFHC1 protein, using in vitro and in vivo models, have uncovered rather unexpected physiological functions for this protein and have led to the proposal of three contrasting and apparently exclusive models for the epileptogenesis (see Fig. 1). This commentary highlights the recent findings on the EFHC1 gene functions and discuses its relevance to the genesis of epilepsy.
EFHC1 as a pro-apoptotic protein: removing the unwanted?
The EFHC1 gene was discovered in the JME critical region 6p12-p11 in the year 2004 and its protein product was found to localize in the soma and dendrites on the neurons.8,14 Ectopic overexpression of EFHC1 resulted in shorter neurites and a fewer branches and the neurons showed signs of degeneration and eventually underwent apoptotic form of cell death.8 Intriguingly, the overexpression of JME associated EFHC1 mutant proteins did not lead to such abnormal neuronal features, suggesting that the mutations might have affected some of the functional properties of EFHC1; one of such functions could be positively regulating the apoptotic process.7,8 The EFHC1 protein harbors an EF-hand domain and this domain has been shown to bind to calcium.8,13 It is known that binding of calcium induces a conformational change in the EF hand domain-containing proteins which, in most cases, result in the changes in the functional property of the target proteins that often catalyze an enzymatic reaction.15 Therefore, the authors tested whether the EFHC1-induced cell death observed in the neuronal cultures is mediated through the calcium channels. Overexpression of EFHC1 in the presence of calcium channel blockers increased the survival rate of neurons, confirming the hypothesis that EFHC1-induced cell death is mediated through calcium signaling.8 Indeed EFHC1 was found to positively regulate activity of calcium channels, and the increased influx of calcium ions could underlie the apoptosis.1,8 Thus, it could be concluded that the JME linked mutations in the EFHC1 might affect the apoptotic process in the brain and that absence of neuronal death could underlie the epileptic symptoms in JME patients.7,8 This conclusion brings in an intriguing question; why should neurons be killed for a brain to be healthy? An answer to this question comes from our understanding of the developmental processes. Programmed cell death – the alternate name for apoptosis, is an important even during development of the embryo and also its central nervous system. Nearly 70% of the developing neurons are known to be eliminated by programmed cell death at various stages for the morpho-genetic events like the neural tube closure.16 Thus loss of EFHC1 might affect this process and brain may end up having additional and unwanted neurons in unwanted places resulting in the hyperexcitable circuits and the epileptic seizures.7 This is the first of the three alternate hypothesis on the role of EFHC1 in JME.
EFHC1 as an axonemal protein: beating for a cause?
The domain organization of EFHC1 protein is almost identical to Rib72 – an axonemal protein of the unicellular flagellate, Chlamydomonas.17 The axoneme represents the inner cytoskeletal compartment of the flagellum or the cilium and provides support to its movement.17 The Rib72 of Chlamy-domonas is thought to function in the structural assembly of the axoneme and in the regulation of flagellar motility.17 Intriguingly, the murine homologue of EFHC1 was found to be abundantly localized to the sperm flagella and tracheal epithelial cilia – the cell types that are known to have axonemal cytoske-leton, suggesting that EFHC1 might be required for the axonemal function in mammalian cells as well.18,19 However, the causal link between ciliary/flagellary function and epilepsy is yet to be established because ciliary/flagellary defects should lead to additional phenotypes (for example, poor fertility, respiratory problems, etc) and such symptoms are not commonly seen in JME patients. Is there a connection between the functions of the cilium and the brain? Yes, indeed; the brain harbors canal-like structure, known as ventricular system, and the cells that line the surface of the canal is characterized by the presence of long cilia. These cells, called the ependymal cells, help in the circulation of the cerebrospinal fluid around the central nervous system, through their rhythmic beating.20,21 Mutations affecting this ependymal flow of the cerebrospinal fluid might result in hydrocephalus - a developmental anomaly associated with the increased intracranial pressure and progressive enlargement of the head in humans.21 A recent study demonstrated that EFHC1 is indeed expressed in ependymal cells, and is localized in cilia.22 While the ciliary structure was unaffected, the ciliary beating was severely impaired in the EFHC1 null mice, suggesting that EFHC1 is required for the normal functions of ependymal cells.23 Intriguingly, the null mice also displayed spontaneous myoclonus and also showed increased susceptibility for induced seizures. Thus, reduction or loss of function of EFHC1 protein results in impaired ciliary function in the ependymal cells and enhanced seizure susceptibility and epileptogenicity. This is the second alternate hypothesis for the role of EFHC1 in JME.
EFHC1 and cell cycle: A neuronal navigator?
Cell biological investigations on the EFHC1 protein led to some unexpected observations. Firstly, EFHC1 was found to associate with the mitotic spindle, and the centrosome.24 Since the subcellular localization of the protein varied during the mitotic cycle, and since only a fraction of the protein gets recruited to the mitotic apparatus, it was suggested that EFHC1 could be a microtubule associated protein, and a potential role for EFHC1 in mitotic cell division was envisaged.24 The EFHC1 gene is expressed maximally in the brain during its embryonic development – a stage where cell division is at maximum, suggesting that EFHC1 could possibly be involved in cell cycle regulation in the developing brain.8,24 What should be the connection between cell cycle and JME? Emerging studies show that neuronal cytoskeletal machinery regulates the cell-cycle of the neuronal progenitors, and that the proliferation of the progenitor cells in the developing cortex is critical for proper neurogenesis, neuronal migration, and axonal wiring.25,26 Since these three events – neurogenesis, neuronal migration, and axonal wiring – are dynamic in nature, and are regulated by microtubule-associated proteins, a causal role for EFHC1 in the proliferation of neuronal progenitors and migration was proposed and tested.27 Firstly, it was shown that overexpression of truncated versions of the EFHC1 in a human kidney cell line resulted in the impaired mitotic spindle defects, and chromosomes misalignments during the metaphase, and apoptosis27 Conversely, RNAi-mediated of knockdown of EFHC1 in the same cell line led to an increase in the mitotic index, suggesting that EFHC1 negatively regulates M-phase progression.27 Secondly, knockdown of EFHC1 in developing cortical region severely affected the neuronal migration. This defect could possibly be due to the fact that the progenitor cells that were devoid of EFHC1 did not exit the cell-cycle and continued to proliferate mitotically.27 Taken together these results have demonstrated that EFHC1 is a regulator of cell division and neuronal migration during development, and defects in this process may underlie some of the symptoms seen in JME. This is the third alternate hypothesis for the role of EFHC1 in JME.
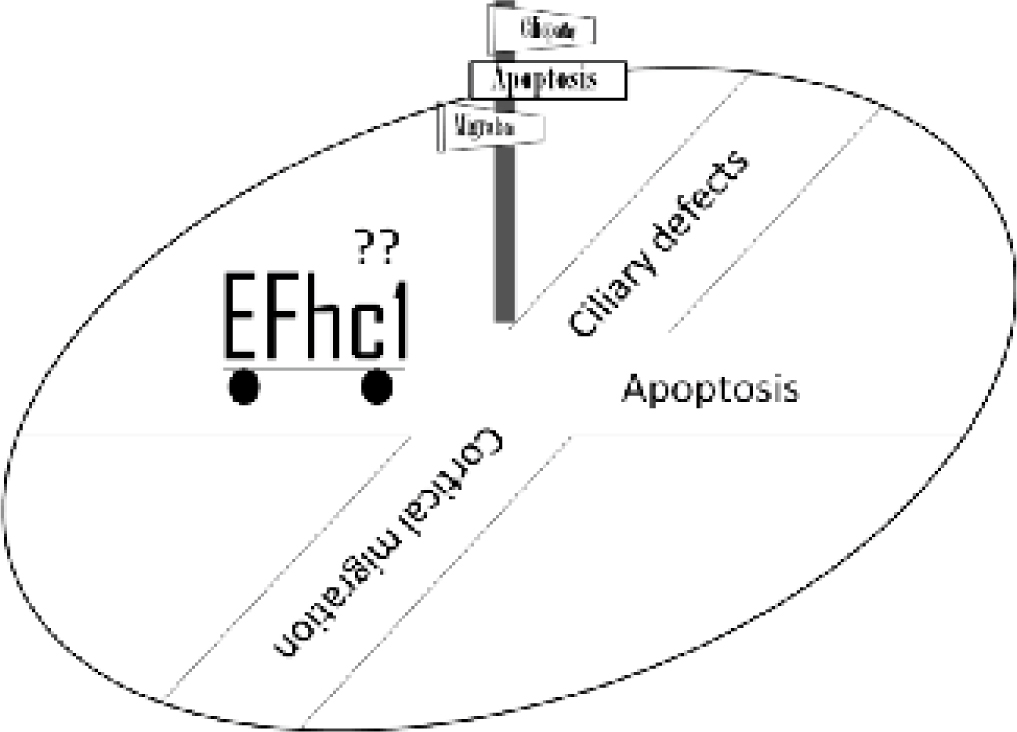
Fig. 1 :Cartoon depicting the direction of three distinct working hypotheses on the role of EFHC1 protein in the etiology of JME. The EFCH1 protein implicated in the proapoptotic function, in the functions of the cilia and in the migration of the neurons. However which one of these three pathways is involved in the epileptogensis is unclear.
EFHC1, the epilepsy gene: where it leads us to?
The positional cloning of genes for monogenic neurological disorders has thrown up a large number of previously uncharacterized genes into the mainstream of neuroscience research. In a way similar to the "reverse genetics" screen in model systems, the functional studies on genes associated with the diseases like JME have discovered novel links between common cellular processes and brain specific disorders. For EFHC1, for example, none of the three proposed mechanisms call for critical role for ion channel, an age old concept that epilepsy results from primary defects in the ion channels. The proposed functions of the EFHC1 proteins – (i) it promotes cell death, (ii) it is involved in ciliary movement, and (iii) it regulate the neuronal differentiation and their migration – may appear to be apparently unconnected. Nonetheless, each of these hypotheses propose the structural changes in brain to be an underlying cause for the epilepto-genesis, and that the JME could be a developmental disorder that manifests in the adolescence. The emerging knowledge on EFHC1 questions the very concept that the JME is one of the idiopathic epilepsies – that is epilepsy without structural lesions in the brain. With growing knowledge in brain development and function, and further studies on the EFHC1 gene, the next few years should witness a significant improvement in our understanding on the cellular functions of EFHC1 protein, and how defects in this process may lead to some of the symptoms seen in JME. Such strides might help us in developing effective strategies for early interventions and therapeutics.
Acknowledgements :
Research work on molecular aspects of epilepsy in the author's laboratory is supported by sponsored research grants from the Department of Science & Technology, Department of Biotechnology, and the Board of Radiation and Nuclear Sciences.
doi : 10.5214/ans.0972-7531.1017202
Subramaniam Ganesh
Deptt of Biological Sciences and
Bioengineering, Indian Institute of
Technology, Kanpur 208016, INDIA
E-mail : sganesh@iitk.ac.in
Tel: 91-512-259 4040
Fax: 91-512-259-4010
References
1. Turnbull J, Lohi H, Kearney JA. Sacred disease secrets revealed: the genetics of human epilepsy. Hum Mol Genet. 2005; 14: 2491-500.
2. Delgado-Escueta AV, Perez-Gosiengfiao KB, Bai D, et al. Recent developments in the quest for myoclonic epilepsy genes. Epilepsia 2003; 44: 13-26 (Suppl 11).
3. Durón RM, Medina MT, Martínez-Juárez IE. Seizures of idiopathic generalized epilepsies. Epilepsia 2005; 46:34-47 (Suppl 9)
4. Hahn A, Neubauer BA. Sodium and potassium channel dysfunctions in rare and common idiopathic epilepsy syndromes. Brain Dev. 2009; 31:515-520.
5. George AL Jr. Inherited Channelopathies Associated with Epilepsy. Epilepsy Curr 2004; 4:65-70.
6. Reid CA, Berkovic S F, Petrou S. Mechanisms of human inherited epilepsies. Prog Neurobiol. 2009; 87: 41-57.
7. Ganesh S, Singh S. Neuronal survival in epilepsy: to die or not to die? J Biosci 2005; 30: 561-566.
8. Suzuki T, Delgado-Escueta AV, Aguan K, et al. Mutations in EFHC1 cause juvenile myoclonic epilepsy. Nat Genet 2004; 36:842-849.
9. Stogmann E, Lichtner P, Baumgartner C, Idiopathic generalized epilepsy phenotypes associated with different EFHC1 mutations. Neurology 2006; 67: 2029-2031.
10. Annesi F, Gambardella A, Michelucci R, et al. Mutational analysis of EFHC1 gene in Italian families with juvenile myoclonic epilepsy. Epilepsia. 2007; 48:1686-1690.
11. Medina MT, Suzuki T, Alonso ME, et al. Novel mutations in Myoclonin1/EFHC1 in sporadic and familial juvenile myoclonic epilepsy. Neurology 2008; 70: 2137-2144.
12. Delgado-Escueta AV. Advances in genetics of juvenile myoclonic epilepsies. Epilepsy Curr 2007; 7:61-67.
13. Murai MJ, Sassonia RC, Zamboni AH, et al. Characterization of the C-terminal half of human juvenile myoclonic epilepsy protein EFHC1: dimer formation blocks Ca2+ and Mg2+ binding to its functional EF-hand. Arch Biochem Biophys 2008;477: 131-138.
14. Léon C, de Nijs L, Chanas G, et al. Distribution of EFHC1 or Myoclonin 1 in mouse neural structures. Epilepsy Res 2010; 88: 196-207.
15. Grabarek Z. Structural basis for diversity of the EF-hand calcium-binding proteins. J Mol Biol 2006; 359: 509-525.
16. De Zio D, Giunta L, Corvaro M, et al. Expanding roles of programmed cell death in mammalian neurodevelopment. Semin Cell Dev Biol 2005; 16: 281-294.
17. Ikeda K, Brown JA, Yagi T, et al. Rib72, a conserved protein associated with the ribbon compartment of flagellar A-microtubules and potentially involved in the linkage between outer doublet microtubules. J Biol Chem 2003; 278: 7725-7734.
18. Ikeda T, Ikeda K, Enomoto M, et al. The mouse ortholog of EFHC1 implicated in juvenile myoclonic epilepsy is an axonemal protein widely conserved among organisms with motile cilia and flagella. FEBS Lett. 2005; 579: 819-822.
19. King SM. Axonemal protofilament ribbons, DM10 domains, and the link to juvenile myoclonic epilepsy. Cell Motil Cytoskeleton 2006; 63: 245-253.
20. Ibañez-Tallon I, Heintz N, Omran H. To beat or not to beat: roles of cilia in development and disease. Hum Mol Genet 2003;12: R27-35.
21. Ibañez-Tallon I, Pagenstecher A, Fliegauf M, et al. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum Mol Genet 2004; 13: 2133-2141.
22. Suzuki T, Inoue I, Yamagata T, et al. Sequential expression of Efhc1/myoclonin1 in choroid plexus and ependymal cell cilia. Biochem Biophys Res Commun 2008; 367: 226-233.
23. Suzuki T, Miyamoto H, Nakahari T, et al. K. Efhc1 deficiency causes spontaneous myoclonus and increased seizure susceptibility. Hum Mol Genet 2009; 18: 1099-10109.
24. de Nijs L, Lakaye B, Coumans B, et al. EFHC1, a protein mutated in juvenile myoclonic epilepsy, associates with the mitotic spindle through its N-terminus. Exp Cell Res 2006; 312: 2872-2879.
25. Shu T, Tseng HC, Sapir T, et al. Doublecortin-like kinase controls neurogenesis by regulating mitotic spindles and M phase progression. Neuron. 2006 Jan 5;49(1):25-39
26. Weimer JM, Anton ES. Doubling up on microtubule stabilizers: synergistic functions of doublecortin-like kinase and doublecortin in the developing cerebral cortex. Neuron 2006; 49: 3-4.
27. de Nijs L, Léon C, Nguyen L, et al. EFHC1 interacts with microtubules to regulate cell division and cortical development. Nat Neurosci 2009; 12: 1266-1274.
(c) Annals of Neurosciences.All Rights Reserved