Annals of Neurosciences, Vol 17, No 3 (2010)
Annals of Neurosciences, Volume 17, Number 3, July 2010
In Vivo application of RNAi to study pain
ABSTRACT
Chronic pain is associated with several disease conditions. The inadequacy of current analgesics to treat chronic pain is the result of a lack of understanding of the mechanisms that mediate pain. RNA interference has emerged in recent years as a new way to evaluate the roles of molecules involved in the pain response. Selective knockout of proteins has proven to be a powerful technique for target validation, but has been limited as a potential therapeutic due to short-lived responses induced by RNAi. The short responses of RNAi illustrate the need for better delivery techniques, which is being addressed by current work to induce RNAi through the cell’s natural mechanisms. In order to gain a better understanding of chronic pain, it will be necessary to evaluate the pain molecules that are expressed as part of an injury induced pain response, which can be modeled by contusion spinal cord injury. RNAi will prove to be an important technique in this work. The present minireview will summarize the work that has been done using RNAi in vivo to study pain and discuss future directions for the use of RNAi to study chronic pain.
KEYWORDS: Chronic pain, RNAi, Pain, Dicer, Spinal cord
Corresponding Author: Daniel K Resnick, MD, MS, E-mail : resnick@neurosurg.wisc.edu
doi : 10.5214/ans.0972-7531.1017310
Introduction
The development of chronic pain is a common sequelae associated with several diseases. The high prevalence of patients suffering from chronic pain highlights the need for effective analgesics therapies. Currently, the two most effective analgesic treatments available are nonsteroidal anti-inflammatory drugs (NSAIDS) and opioids, both of which have serious side effects and often relieve pain only transiently. Pain research has identified several molecules important in mediating nociception that cannot be targeted by conventional analgesics. Recent advances in molecular biology have enabled the selective knockdown or suppression of these target gene products in animal models. One of the most promising molecular techniques to emerge in pain research is RNA interference (RNAi).
RNA interference is a mechanism through which double stranded RNA (dsRNA) is processed by the cell and used to interrupt formation of gene products by degrading mRNAor suppressing its translation.1 RNAi is a naturally occurring process in plants and invertebrates, where it was originally discovered and referred to as co-suppression and quelling respectively2,3. The term RNA interference was coined by Fire and Mello when they described this process in Caenorha habditis elegans.4 RNAi may be mediated by small interfering RNA (siRNA), which leads to selective mRNA degradation or by microRNA (miRNA), which suppresses mRNA translation.5 RNAi can be induced in mammals by dsRNA (>30 bp), expression vectors leading to transcription of short hairpin RNA (shRNA), or by administration of siRNA directly.6 The present review will discuss different methods to induce RNAi in vivo for application in pain research.
Mechanism of RNAi
RNAi degrades mRNA through a complex of proteins known as the RNA-lnduced Silencing Complex (RISC). The first step in forming the RISC is the production of siRNA through the activity of the type III ribonuclease Dicer.7 Dicer cleaves its substrate to form double stranded siRNA that is 21-23 nucleotides long with 3’ overhangs of 2-3 nucleotides on each strand. Dicer substrates include dsRNA, shRNA, or synthetic Dicer substrate siRNA (DsiRNA). Dicer cleavage can be circumvented by direct application of synthetic siRNA that is already 21 bases long (Figure 1). Following cleavage. Dicer and 21-nucleotide siRNA come into contact with another enzyme, Argonaute 2 (Ago2), which along with HIV-Transactivating Response RNA-Binding Protein (TRBP) form the RISC-Loading Complex (RLC). Ago2 cleaves the siRNA passenger strand and release it from the RLC.8,9,10 When only the guide strand of siRNA remains, the RISC is formed. The RISC binds mRNA that is complementary to the siRNA guide strand and directs mRNA breakdown.
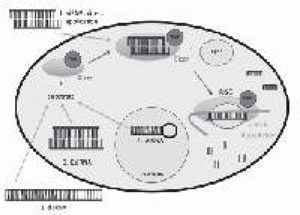
Fig. 1. Dicer substrates that induce RNAi. 1. 21-nucleotide siRNA can be delivered directly to spinal cord in transfection agents including cationic lipid, polyethyleneimine, and nanopartides. 2. Double stranded RNA (usually >30 bp) can be cleaved by Dicer to induce RNAi. 3. Dicer substrate siRNA (DsiRNA) is a 27-nucletide sequence that can induce RNAi. 4. Short hairpin RNA (shRNA) can be delivered to cells using herpes simplex virus or lentivirus. Once transcribed, shRNA is transported out of the nucleus and can become a Dicer substrate.
Direct siRNA delivery
One of the biggest challenges in using RNAi in pain research is delivery of siRNA to the CNS in sufficient concentrations. This obstacle exists because siRNA by itself does not cross the blood brain barrier (BBB) and is degraded in the blood by endonucleases. Intravenous or oral administration is, therefore, inadequate to achieve desired protein knockdown.1,6 The use of transfection agents and intrathecal delivery has enhanced siRNA uptake by target tissues in recent studies.
One of the earliest pain studies using siRNA in vivo was targeted against the P2X3 receptor, an ATP-gated cation channel which signals nociception.11 Naked P2X3 siRNA was delivered intrathecally to L1 at a concentration of 400 g/day continuously for up to 7 days. P2X3 siRNA administration resulted in lower agonist-induced mechanical hyperaglesia and tactile allodynia than vehicle and mismatch siRNA treated controls. siRNA treated animals with neuoropathic pain (partial sciatic ligation model) also showed lower tactile allodynia and mechanical hyperalgesia than neuropathic control animals. These behavioral effects were due to knockdown of the P2X3 receptor, which was quantified as a 40% reduction in P2X3 mRNA in dorsal root ganglia (DRG) of non-neuropathic siRNA treated animals. In addition, P2X3 siRNA was shown to be more potent than P2X3 antisense oligonucleotides (ASO). Antisense oligonucleotides are synthetic single strand stretches of oligonucleotides that bind to complementary mRNA and prevent translation.12 The authors had previously established that P2X3 ASO were effective in reducing P2X3 mRNA levels and lowering mechanical hyperalgesia but found in the current study that an equivalent dose of siRNA gave a stronger effect.13
In a subsequent study, siRNA was used to target the delta opioid receptor (DOR), which mediates antinocicpetion.14 DOR siRNA was delivered once daily for three consecutive days to the lumbar region of the spinal cord through an implanted intrathecal catheter. siRNA was delivered using a cationic lipid transfection agent (i-Fect™, Neuromics), which enabled the use of a much lower effective dose of siRNA than used for naked siRNA delivery in the previous study. 2g of DOR siRNA delivered in 10 l of i-Fect™ transfection agent proved to be effective in reducing DOR mRNA levels in the lumbar spinal cord (62% reduction) and DRG (38% reduction). In addition, agonist-induced anti-nociception was reduced 24 hours following the last dose of siRNA compared to vehicle and mismatch controls. However, 72 hours following the last siRNA dose, there was no significant difference in antinocicpetion between groups illustrating the temporary effect of RNAi using this delivery method.
The i-Fect™ transfection reagent was also used in recent studies to evaluate the role of ion channels in mediating neuropathic pain. siRNA directed against Nav1.8, a Na channel subtype that is insensitive to tetrodotoxin was able to effectively reduce Nav1.8 mRNA by 31 % in L4 and L5 DRG cells.15 100 I (concentration of. 2g siRNA/1 I of i-Fect™) of siRNA solution was delivered once daily for four consecutive days through an implanted epidural catheter at L4 to target lumbar DRG cells. By the third day of siRNA treatment, animals that had previously undergone chronic constriction injury had significantly lower pain responses than vehicle treated animals with the same injury. This was measured as an increased paw withdrawal time in a mechanical allodynia assay, which peaked 24 hours following the last bolus of siRNA.
In another ion channel study, siRNA was directed against Cav1.2, a voltage-gated calcium channel upregulated after spinal nerve ligation. siRNA directed against this channel was effective in reducing mechanical allodynia 24 hours following the last dose of siRNA. Cav1.2 siRNA was delivered intrathecally daily for 4 days just above the lumbar region of the spinal cord through an implanted catheter at a dose of 2 g siRNA per 10 l of i-Fect™.16
In addition to the cationic lipid i-Fect™, other transfection reagents have been combined with siRNA to achieve effective target knockdown using low doses of siRNA. Tan and colleagues utilized the cationic polymer polyethyleneimine (PEI) to deliver siRNA directed against the NMDA receptor subunit NR2B.17 NMDA receptor activation in the spinal cord produces hyperalgesia, suggesting a role for the receptor in pain signaling.18 The NR2B subunit was chosen for target knockdown because of its previously established localization in the dorsal horn of the spinal cord).19 NR2B siRNA was delivered intathecally to the lumbar region through an implanted catheter in a ratio of. 18 l of PEI per 1 g of siRNA. Maximal reduction of NR2B was achieved three days following siRNA delivery with 84% inhibition of NR2B mRNA for a 5 g siRNA dose. NR2B mRNA levels remained significantly lower in siRNA treated animals than controls 14 days following siRNA delivery but were indistinguishable from controls by 21 days post-delivery. To assess pain, animals were given 1% formalin injections, a model for spontaneous pain. siRNA treated animals showed lower pain responses than controls 7 and 14 days following siRNA delivery using the formalin assay. Pain responses were not significantly different 3 days following treatments. These results indicate that the RNA-polymer complex used in this study had a longer latency for target mRNA reduction than the cationic lipid transfection agent i-Fect™ did for the DOR (3 days vs 1 day). Effects also appear to be longer lasting with this method as a lower formalin-induced pain response was seen 14 days following treatment, whereas the antinociception response mediated by the DOR was indistinguishable between the siRNA treated group and controls 3 days following delivery.
Another effective method for intrathecal delivery of siRNA is the use of nanoparticles. To assess the function of different muscarinic acetylcholine receptors (mAChRs) nanoparticles were prepared from the linear polysaccharide chitosin and complexed with siRNA (nanoparticle preparation described previously.20 Three of the five different subtypes of mAChRs were targeted in the study because of their location in the dorsal horn established by prior work.21 Chitosan-siRNA nanoparticles were prepared with siRNA sequences directed against M2, M3, and M4 mAChR subtypes at a concentration of 1 g siRNA per 1 I of chitosan solution. Intrathecal injection of chitosan-siRNA nanoparticles to the lumbar region was done once daily for 3 consecutive days through an implanted catheter at a dose 5 g of siRNA.22 3 days following the final injection, mRNA was reduced in the lumbar DRG and spinal cord for M2, M3 and, M4 subtypes (~50-60%). This demonstrates the effectiveness of chitosan nanoparticles for siRNA delivery in vivo. Thermal paw withdrawal latencies were measured following administration of the agonist muscarine to assess the antinociception responses of the receptors. Agonist-induced antinocipetion was diminished 3 days following the last siRNA injection for M2 and M4 subtypes suggesting that these subtypes are important for the muscarine analgesic response.
Dicer substrate siRNA
In all of the previous studies described, RNAi was induced by delivery of siRNA directly to the spinal cord. Substrate cleavage by the Dicer endonuclease was not necessary to induce RNAi because 21 -nucleotide siRNA was already prepared and delivered in isolation or in a transfection agent. Recent work has shown that delivery of a 27-nucleotide sequence of siRNA may be a more potent mechanism to induce RNAi in vivo. In this study, a 27-nucleotide sequence of siRNA that could act as a substrate for Dicer was prepared to target the neurotensin receptor-2 (NTS2).23 NTS2 is a G protein-coupled receptor expressed in the spinal cord that results in analgesia when stimulated.24,25 NTS2 DsiRNA was injected twice (24 hours between injections) intrathecally between L5-L6 vertebrae at a dose of 1 g of DsiRNA delivered in 10 l of saline and i-Fect™ transfection agent. 24 hours after the last injection, DsiRNA treated animals showed reduction in NTS2 mRNA in the lumbar DRG (82%) and lumbar spinal cord (62%). Reduction in NTS2 mRNA was accompanied with expected behavioral responses using a tail flick test. Vehicle treated animals as expected showed an increase in tail flick response latencies when treated with the NTS2 agonist JMV-431. This aninociceptive effect was lost in DsiRNA treated animals 24 hours after the last injection. This persisted up to 4 days after the last dose of DsiRNA was delivered. Although the effects of the present delivery method were not sustained as long as the PEI delivery technique (14 days), using DsiRNA in the cationic lipid i-Fect™ gave longer lasting effects than siRNA delivered in i-Fect™ (4 days vs less than 3 days).
In addition to the use of intrathecal catheters to deliver siRNA to the spinal cord, siRNA has been successfully delivered through injection by a transcutaneous approach. Miletic and colleagues delivered siRNA directed against HomeM proteins, which area class of post-synaptic density proteins thought to be important in the development of the pain response.26 101 of 1 M Homerl siRNA was injected into the spinal cord between vertebral levels L5 and L6 using a 25G needle. The needle was inserted using a transcutaneous approach described in previous work.27,28 Pretreatement of Homerl siRNA was effective in altering Homerl protein expression following chronic constriction injury (CCI) of the sciatic nerve. Both upregulation of the Homerl a protein and downregulation of Homerl b/c protein subclasses, which normally follow CCI were inhibited by Homerl siRNA pretreatment. In addition to altering Homerl protein expression, Homerl siRNA treatment also attenuated thermal hyperalgesia following CCI providing behavioral evidence for the importance of Homerl proteins in mediating the pain response. These results confirm that the transcutaneous approach is another effective technique for siRNA delivery in vivo. The transcutaneous approach for siRNA delivery was further validated by this group in a more recent study in which they were able to knock down Shankl protein levels (another PSD protein involved in developing the pain response).29
Short hairpin RNA
Another molecule known to induce RNAi is short hairpin RNA (shRNA). ShRNA is double stranded RNA that contains a loop of base pairs and is longer than 21-nucelotide siRNA. ShRNA is transcribed in the nucleus of the cell and subsequently transported to the cytoplasm where it can serve as a substrate for Dicer, which cleaves out the hairpin loop and forms siRNA that is 21-23 nucleotides long.5 One drawback of using synthetic 21-nucleotide siRNA to induce RNAi is that the effects are short-lived. Providing substrates for Dicer acts upstream of direct siRNA delivery and is closer to the cell’s natural RNAi mechanism. New efforts to induce RNAi in vivo have focused on mimicking more of a natural response in order to get a more potent effect as was seen using DsiRNA.30
Two studies to date have used shRNA in vivo to target pain molecules. The first used a replication-defective Herpes Simplex Virus (HSV-1) vector to deliver shRNA.31 ShRNA sequences were designed to target the transient receptor potential vanilloid 1 (TRPVI)gene, known to play an important role in nociceptive signaling.32Up to 71 of HSV-1 vector was injected into the sciatic nerve using different promoter systems to express TRPV1 shRNA. The vector was successfully transfected into lumbar DRG through retrograde transport. One promoter system (HSV-U6shTrpv1) effectively reduced TRPV1 mRNA in L4 DRG by 53.6% as measured by RT-PCR. Interestingly, the investigators were also able to achieve TRPV1 protein knockdown (59%) using a miRNA promoter system (HSV-GFP-miR-TRPV1), an original result.
Transfection of shRNA was also achieved using a lentivirus vector in a later study. Lentivirus constructs were designed to express shRNA directed against the DREAM gene, which codes for a multifunctional protein that has been implicated in pain signaling. Intrathecal administration of the lentivirus vector was successful in reducing DREAM mRNA in the lumbar spinal cord.33 This study provides another method for transfecting shRNA to target pain molecules in vivo.
Conclusion
RNAi has proven to be a powerful technique for target validation in recent years. In the context of pain research, it has provided a way to evaluate the roles of several molecules that are involved in the pain response that cannot be targeted by conventional treatments. The biggest obstacle in using RNAi technology in vivo has been delivery to the CNS. Delivery has been improved through the use of transfection agents such as the cationic lipid i-Fect™, polyethyleneimine, and chitosan-nanoparticles.These agents have enabled the delivery of siRNA directly to the spinal cord through intrathecal injection but the effects of direct delivery have been relatively short-lived. Because of this, recent work has focused on induction of RNAi through the use of Dicer substrates such as DsiRNA and shRNA in order to achieve longer lasting effects. This work is still in its infancy however.
Behavioral responses assessed using RNAi have been largely agonist-induced responses or sciatic ligation model responses. There has been little done to assess how RNAi might affect pain molecules that are differentially expressed following spinal cord injury (SCI). In order to better understand how chronic neuropathic pain is mediated it will be necessary to target pain molecules with RNAi that are induced following SCI. Neuropathic pain induced by contusion spinal cord injury (cSCI) may provide a good model system for evaluating this issue. Several studies have shown differential expression of proteins following cSCI, which may be good targets for RNAi.34,35,36 The best technique to induce RNAi in a spinal cord injury model remains to be determined.
Competing interests - None
Received Date : 22 June 2010
Revised Date : 10 August 2010
Accepted Date : 12 August 2010
References
1. Tan PH, Yang LC, Ji RR. Therapeutic potential of RNA interference in pain medicine. Open Pain J 2009;2:57-63.
2. Napoli C, Lemieux C, Jorgensen R. Introduction of a Chimeric Chalcone Synthase Gene into Petunia Results in Reversible Co-Suppression of Homologous Genes in trans. Plant Cell 1990;2:279-289.
3. Romano N, Macino G. Quelling: transient inactivation of gene expression in Neurospora crassa by transformation with homologous sequences. Mol Microbiol 1992;6:3343-3353.
4. Fire A, Xu S, Montgomery MK, et al. Potent and specific genetic interference by double-stranded RNA in Caenorhabditis elegans. Nature 1998;391:806-811.
5. Rao DD, Vorhies JS, Senzer N, et al. siRNA vs. shRNA: similarities and differences. Adv Drug Deliv Rev 2009;61:746-759.
6. Rohl T, Kurreck J. RNA interference in pain research. J Neurochem 2006;99: 371-380.
7. Bernstein E, Caudy AA, Hammond SM, et al. Role for a bidentate ribonuclease in the initiation step of RNA interference. Nature 2001;409:363-366.
8. Matranga C, Tomari Y, Shin C, et al. Passenger-strand cleavage facilitates assembly of siRNA into Ago2-containing RNAi enzyme complexes. Cell 2005;123: 607-620.
9. Preall JB, Sontheimer EJ. RNAi: RISC gets loaded. Cell 2005;123:543-545.
10. Rand TA, Petersen S, Du F, et al. Argonaute 2 cleaves the anti-guide strand of siRNA during RISC activation. Cell 2005;123:621-629.
11. Barclay J, Patel S, Dorn G, et al. Functional down regulation of P2X3 receptor subunit in rat sensory neurons reveals a significant role in chronic neuropathic and inflammatory pain. J Neurosci 2005;22:8139-8147.
12. Caruso G, Caffo M, Raudino G, et al. Antisense oligonucleotides as innovative therapeutic strategy in the treatment of high-grade gliomas. Recent Pat CNS Drug Discov 2010;5:53-69.
13. Dorn G, Patel S, Wotherspoon G, et al. siRNA relieves chronic neuropathic pain. Nucleic Acids Res 2004;32:49.
14. Luo MC, Zhang DQ, Ma SW, et al. An efficient intrathecal delivery of small interfering RNA to the spinal cord and peripheral neurons. Mol Pain 2005;1:29.
15. Dong XW, Goregoaker S, Engler H, et al. Small interfering RNA-mediated selective knockdown of Na(V)1.8 tetrodotoxin-resistant sodium channel reverses mechanical allodynia in neuropathic rats. Neuroscience 2007;146:812-821.
16. Fossat P, Dobremez E, Bouali-Benazzouz R, et al. Knockdown of L calcium channel subtypes: differential effects in neuropathic pain. J Neurosci 2010;30:1073-1085.
17. Tan PH, Yang LC, Shih HC, et al. Gene knockdown with intrathecal siRNA of NMDA receptor NR2B subunit reduces formalin-induced nociception in the rat. Gene Ther 2005;12:59-66.
18. Meller ST, Dykstra C, Gebhart GF. Production of endogenous nitric oxide and activation of soluble guanylate cyclase are required for N-methyl-D-aspartate-produced facilitation of the nociceptive tail-flick reflex. Eur J Pharmacol 1992;214:93-96.
19. Boyce S, Wyatt A, Webb JK, et al. Selective NMDA NR2B antagonists induce antinociception without motor dysfunction: correlation with restricted localisation of NR2B subunit in dorsal horn. Neuropharmacology 1999;38:611-623.
20. Katas H, Alpar HO. Development and characterisation of chitosan nano-particles for siRNA delivery. J Control Release 2006;115:216-225.
21. Hoglund AU, Baghdoyan HA. M2, M3 and M4, but not M1, muscarinic receptor subtypes are present in rat spinal cord. J Pharmacol Exp Ther 1997;281:470-477.
22. Cai YQ, Chen SR, Han HD, et al. Role of M2, M3, and M4 muscarinic receptor subtypes in the spinal cholinergic control of nociception revealed using siRNA in rats. J Neurochem 2009;111:1000-1010.
23. Dore-Savard L, Roussy G, Dansereau MA, et al. Central delivery of Dicer-substrate siRNA: a direct application for pain research. Mol Ther 2008;16:1331-1339.
24. Maeno H, Yamada K, Santo-Yamada Y, et al. Comparison of mice deficient in the high- or low-affinity neurotensin receptors, Ntsrl or Ntsr2, reveals a novel function for Ntsr2 in thermal nociception. Brain Res 2004;998: 122-129.
25. Dobner PR. Neurotensin and pain modulation. Peptides 2006;27:2405-2414.
26. Pearce SM, Ramsey MA, Miranpuri GS, et al. Regulation and function of matrix metalloproteinases in nervous system injury and Neuropathic pain. Annals of Neurosciences 2008;15:94-105.
27. Mestre C, PelissierT, Fialip J, et al. A method to perform direct transcutaneous intrathecal injection in rats. J Pharmacol Toxicol Methods 1994;32:197-200.
28. Miletic G, Miletic V. Loose ligation of the sciatic nerve is associated with TrkB receptor-dependent decreases in KCC2 protein levels in the ipsilateral spinal dorsal horn. Pain 2008;137:532-539.
29. Miletic G, Dumitrascu CI, Honstad CE, et al. Loose ligation of the rat sciatic nerve elicits early accumulation of Shank1 protein in the post-synaptic density of spinal dorsal horn neurons. Pain 2010;149:152-159.
30. Cullen BR. RNAi the natural way. Nat Genet 2005;37:1163-1165.
31. Anesti AM, Peeters PJ, Royaux I, et al. Efficient delivery of RNA Interference to peripheral neurons in vivo using herpes simplex virus. Nucleic Acids Res 2008;36:86.
32. Tominaga M, Tominaga T. Structure and function of TRPV1. Pflugers Arch 2005;451:143-150.
33. Cain JH, Boggott C, Tilghman JI, et al. Recent developments in the study of spinal cord injury and neuropathic pain. Annals of Neurosciences 2007;14:96-107.
34. Hasbargen T, Ahmed MM, Miranpuri G, et al. Role of NKCC1 and KCC2 in the development of chronic neuropathic pain following spinal cord injury. Ann N Y Acad Sci 1198:168-172.
35. DomBourian MG, Turner NA, Gerovac TA, et al. B1 and TRPV-1 receptor genes and their relationship to hyperalgesia following spinal cord injury. Spine (Phila Pa 1976) 2006;31:2778-2782.
36. Rajpal S, Gerovac TA, Turner NA, et al. Antihyperalgesic effects of vanilloid-1 and bradykinin-1 receptor antagonists following spinal cord injury in rats. J Neurosurg Spine 2007;6:420-424.
37. Cramer SW, Baggott C, Cain J, et al. The role of cation-dependent chloride transporters in neuropathic pain following spinal cord injury. Mol Pain 2008;4:36.
(c) Annals of Neurosciences.All Rights Reserved