Annals of Neurosciences, Vol 16, No 2 (2009)
Annals of Neurosciences, Volume 16, Number 2, April 2009
Age related macular degeneration - advances and trends
ABSTRACT
Age related macular degeneration (AMD) is the most common cause of visual impairment and is characterized by drusen formation, geographic atrophy and gradual loss of vision. It is a frightening disease that destroys the macula, the central part of retina, severely regimenting a person’s normal sight. The utility of mouse models with features of AMD along with its recently reported association with complement factor H-gene, and TLR4 genes strongly suggest the importance of inflammatory mediators and complement in the pathogenesis of this disease. Current treatment modalities include photodynamic therapy and photocoagulation, however, their efficacy is limited. The animal models that faithfully replicate the features of human AMD are useful platforms in validating new therapies. This not only provides new insights for preventative and restorative approaches in AMD in future but also advances our understanding of cardiovascular and neurodegenerative disorders.
KEYWORDS: Macular degeneration. Risk Factors, Genes responsible for AMD, Genetic predisposition, Animal models, Therapeutic approaches.
Corresponding Author: Akshay Anand, E-mail: akshay1anand@rediffmail.com, Tel. : +91-172-2756090
doi: 10.5214/ans.0972.7531.2009.160208
Introduction
Any impairment in vision severely limits the ability to maintain a good quality of life. Age-related macular degeneration (AMD) is a leading cause of irreversible blindness in people over 60 years of age in many industrialized countries. It is the principal cause of irreversible, registered legal blindness on three continents1. It is characterized by the progressive degeneration of the retina, RPE and underlying choroid (the highly vascular tissue beneath the RPE) which results in severe vision loss. The earliest clinically visible abnormality in AMD is the accumulation of drusen (lipoproteinaceous deposits) in the extracellular matrix between the RPE and the choroid. The affected portion of the eye is the centre of the retina, called the macula, which contains the highest concentration of cone photoreceptors and is responsible for central vision. Although the majority of AMD cases are the less-severe dry form, ~10- 20% of these patients develop wet, or exudative, AMD. The dry form is characterized by geographic atrophy, the formation of pigmented yellow deposits (called drusen) and the gradual loss of central vision, whereas the wet form of AMD manifests with rapid development of choroidal neovascularization, subretinal membrane formation and photoreceptor atrophy, leading to blindness. CNV is characterized by bleeding or fluid leakage and is usually preceded by drusen formation. Drusen are amorphous deposits that form between the retinal pigment epithelium (RPE) and Bruch’s membrane and comprise immune deposits, lipofuscin and complement. It is the spatiotemporal confluence of these drusen that partly contributes to vision loss.
It is also interesting to note that age-related disorder shares many mechanisms common to atherosclerotic plaque development and drusen formation in glomerular nephritis 2. Drusen deposition is a significant risk factor for progression to CNV, the exudative hallmark of late AMD, and vision loss. Existing animal models attempting to simulate AMD through high-fat diets and phototoxicity, senescence acceleration or candidate gene manipulation1,3 do not fully replicate the clinical, histologic and angiographic features of the human condition.
Recent evidence suggests that complement activation and immune complex deposition occurs in eyes of humans with AMD. In other immune complex deposition disorders, it has been postulated that these proteins serve as an inflammatory focus by inciting macrophage recruitment through Fc and complement receptor binding, triggering humoral activation and phagocytosis. Dry AMD is much more common than wet AMD, but the wet form occurs in about 10% of these cases. Dry AMD is characterized by progressive apoptosis of cells in the epithelial layer, geographic atrophy in the overlying photoreceptor cells and in the underlying cells in the choroidal capillary layer4. Wet AMD looks quite different in this form with proliferating blood vessels hemorrhage that exude serous fluid under the retina. These new vessels usually form a neovascular membrane that originates from the choroids and penetrates Bruch’s membrane into the subretinal space5. Current therapeutic efforts and clinical trials are directed towards hailting the growth of the neovascular membrane in wet AMD, using angiogenesis inhibitors such as anti-VEGF or thermal lasers. Despite considerable research, little is known about the events that trigger AMD. Drusen deposition precedes both the dry and wet forms and is an independent risk factor for the development of a neovascular membrane6. Drusen are considered by some to be the residue of undigested material from dysfunctional phagocytic cells in the epithelial layer. They seem to consist of general biological effluvia cholesterol-rich lipids, various proteins and the like. In particular, oxidized lipoproteins have been identified in these structures7 supporting the notion that they arise through oxidative damage. They also contain potential macrophage chemoattractants, such as complement components and immunoglobulins.8
AMD emerges during a preclinical, asymptomatic phase in which waste material (drusen) accumulates in the space between the basement membrane (Bruch’s membrane) and the epithelial layer. Bruch’s membrane separates the retina from the choroids, the vascular layer of the eye wall that lies between the retina and the sclera. The epithelial layer, dubbed the retinal pigmented epithelial layer is crucial for photoreceptor health. Cells in this layer recycle visual pigment (rhodopsin), phagocytose photoreceptor tips daily as part of rod and cone regeneration and transport fluid across the membrane to the choroids to help prevent detachment of the neural retina. The metabolic demands on the retinal epithelium are considerable as phagocytosis of photoreceptor tips occurs each morning on awakening for 30 to 60 minutes. Central vision vanishes when cells in this epithelial layer cease to function properly, causing photoreceptor degeneration.
The clinical studies show a correlation between drusen formation and autofluorescence (as seen through a scanning laser ophthalmoscope9), an indirect measure of the accumulation of pellet-like deposits in the retinal epithelial cell layer. These deposits, called lipofuscin granules, are partially degraded cell membranes in lysosomes, and accumulate in cells that endocytose oxidized lipoproteins but have no mechanism to adequately dispose of them. This deficiency occurs in professional lipoprotein scavengers10, such as the aging macrophages in atherosclerotic lesions (foam cells) and in nonprofessional phagocytes, such as neurons in the central nervous systems and cells in the retinal epithelial layer. Defects in this housekeeping function in retinal epithelial cells and in resident choroidal macrophages may contribute to drusen accumulation with age11. In addition to its well-known pro-inflammatory talents, MCP-1 may also have an immunomodulatory role. For instance, it seems to shift the balance of the T-cell immune response towards T-helper type 2 by promoting production of the cytokine interleukin-4 (IL-4), and by suppressing the T-helper type 1 associated cytokine IL-1212 cells of the blood retinal barrier, including those of the retinal pigmented epithelial layer, constitutively produce MCP-1 and can secrete high levels of the molecule after exposure to other cytokines and chemokines such as IL MCP-1 might help coordinate the turnover of resident macrophages and surveillant, immature and surveillant, immature dendritic cells, both of which form extensive cellular networks in the choroids that intimately contact with the epithelial cell layer13.
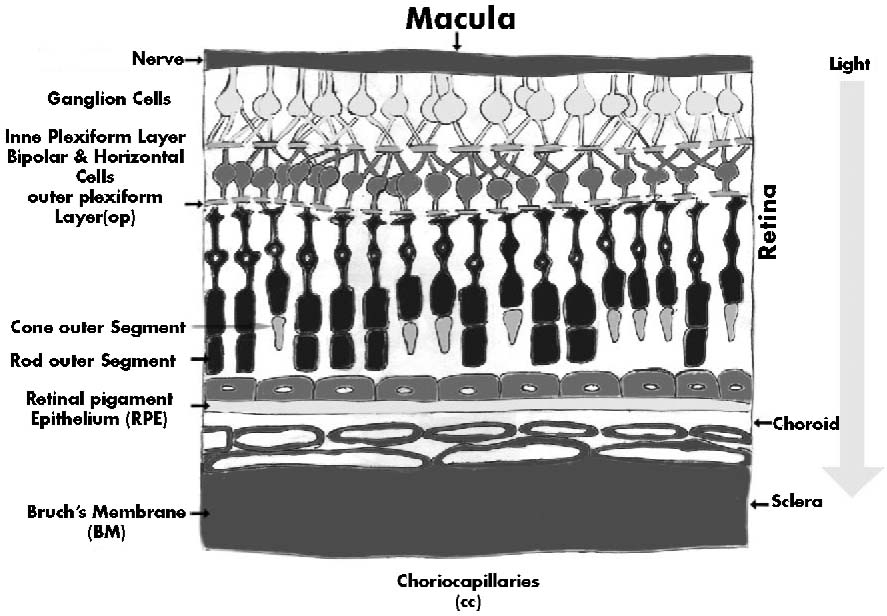
RPE-the strategic player
The neuroectodermally-derived RPE is a major component of the blood-retina barrier lying adjacent to Bruch’s membrane, of which the decreased collagen solubility with age is proposed to contribute to debris accumulation14. AMD is partially characterized by the disruption of interactions of the epithelium with the neural retina and the underlying choroidal vasculature. Retinal function is significantly dependent on the health of RPE cells, which produce useful cytokines and maintain the homeostasis of essential photopigments. In the normal retina, the RPE performs the functions of barrier, macrophage and neuroprotective cell layer. The RPE cell layer forms a polarized and selectively permeable barrier and controls shape15, ion flux16,17 and cell signalling18. The basal surface of the RPE sits on Bruch’s membrane, which is a complex extracellular matrix thought to be the product of both RPE cells and the choroidal vasculature, similar in function to astroglia in the central nervous system (CNS)19 . The cells lie in a monolayer coupled by a variety of cell-cell junctions between RPE and choroidal endothelial cells20. The apical surface of the RPE ensheathes the photoreceptors in the adult retina and supports the renewal of their outer segments. Signalling between the retina and the RPE is complex. RPE apical membranes contain ion pumps, lectin affinity sites, transporters for water and neuroactive peptides and receptors for catecholamines and sugars. The macrophage capacity of RPE cells is believed to support the maintenance of photoreceptors and the clearance of the complement 5 (C5) and IgG deposits that comprise drusen21. It is unclear whether the endogenous macrophages that accumulate in the choroids of patients with AMD serve protective or destructive functions. Although they might aid drusen clearance22, they could also promote CNV23, similar to their proposed role in a laser-injury mouse model 24,25 . These findings suggest that macrophages can serve both pro- and anti-inflammatory capacities and that the dysregulation of clearance functions might facilitate drusen formation and disease progression. These drusen might lead to disturbances in spatiotemporal sensitivity, colour contrast and central vision. Most of these cells are post-mitotic and last for the duration of a human life. In this state, the cells make and secrete retino-protective and anti-angiogenic factors, such as pigment-epithelium-derived factor (PEDF)26,27 and fibroblast growth factor (FGF-2)28. They also express Fas ligand (FasL), and together these factors are reported to control the growth and development of new subretinal vessels, which can damage vision29; however, their role in AMD has not been conclusively established30.
Melanin is another principal pigment in RPE that accumulates only in the nondividing cells of the RPE layer. Mature melanin is known to act as an efficient antioxidant which scavenges free radicals and protects the eye from stray light32,33. White mice are, therefore, expected to be more susceptible to neovascularization than black mice34. When RPE cells are injured by mechanical or thermal damage, the cells respond with regeneration, fibrovascular proliferation and the mobilization of melanin pigment35,36. Lipofuscin, which is chiefly composed of the fluorophore A2E (N-retinylidene-Nretinylethanolamine), accumulates as a hydrophobic component of RPE in AMD-affected eyes and is believed to originate from the incomplete lysosomal digestion of shed photoreceptor outer segments, which increases proportionately with age37,38. Thus, the preservation of RPE functions and compensation for age-related changes are central to the prevention of AMD. Although newer medical approaches, such as PDT, the injection of angiostatic steroids39 or laser photocoagulation, attempt to inhibit the growth of abnormal blood vessels from the choriocapillaries into the outer retina, there are greater challenges for the development of novel gene and cell therapies to treat the causes of this retinal disease.
Genetic predisposition
Studies on the molecular composition of drusen have implicated inflammation, and particularly local activation of the alternative pathway (AP) of the complement cascade in the retina, in the pathogenesis of AMD40. Furthermore, strong evidence for a role of complement in this disease derives from an independent line of research which showed that variants in the complement factor Η (CFH) gene are significantly associated with an increased risk for AMD in Caucasian populations41. These genetic studies were recently extended by the observation that polymorphisms in other complement genes, notably those coding for factor B-complement component C2 (BF-C2) and complement C3 (C3), are also associated with AMD42,43. There have been studies where LOC387715 and HTRA1 polymorphisms have been associated with a higher risk of exudative AMD in northern Chinese although there are also reports where no association of CFH Y402H with exudative AMD have been suggested44. AMD susceptibility, like any disease, is linked to both genetic and environmental factors, although its precise etiology remains elusive. Reported risk factors include ocular pigmentation, dietary factors, positive family history for AMD, smoking, and several gene mutations such as ATP-binding cassette transporter protein 4 (ABCA4), apolipoprotein Ε {APOE), and fibulin-5 (FBLN5)45-50. Moreover, genome-wide linkage studies have successfully identified several major chromosomal regions including 1q31 and 10q26 51-55. Furthermore, studies of Hong Kong Chinese and Caucasian population have identified SNP rs11200638:G>A in the promoter of high-temperature requirement A-1 (HTRA1) gene, approximately 6.1 kb downstream of rs10490924 in LOC387715 on chromosome 10q26, to be associated with increased risk of AMD56,57. There are studies to suggest that theTyr402His coding allele is associated with wet AMD in the Israeli population58. With several loci being implicated in the pathogenesis of AMD, another group has identified that one of the genetic variants (251A/T), which is associated with a gene that boosts the production of interleukin 8 (known as IL-8), was significantly more common among the patients with AMD. This held true even after taking account of age, sex, weight, and smoking, which is a known risk factor for AMD. Klein et a/59 have reported a genome-wide screen of 96 cases and 50 controls for polymorphisms in the complement factor Η gene (CFH) that are associated with AMD. In individuals that are homozygous for the CFH risk allele, the likelihood of AMD is increased by a factor of 7.4. This report tends to prove the ‘common-gene-common-disease’ hypothesis and endorses the strong association of an inflammatory role in the disease. The analysis of tyrosine-histidine polymorphisms, primarily involved in CFH, should also be studied in other populations. Not withstanding this report, the genetic basis of AMD as a single-gene disorder is uncertain. The genes encoding chemokine ligand-2 (Ccl2) and its cognate receptor, chemokine receptor-2 (Ccr2), have also evoked considerable interest after a new animal model for AMD was developed. Gene hunters are screening the Ccl2 and Ccr2 genes in patients with AMD through various multi-centre studies. It is unclear if the mirage of Ccl2 or Ccr2 polymorphisms would provide any causal link to AMD60. However, current studies among Caucasian populations and US population did not provide any evidence of an association between TLR4, CCR2, and CCL2 and AMD, which implies that the common genetic variation in these genes does not play a significant role in the etiology of AMD61 although suggested by several other studies. CFH polymorphism Tyr402His among Indian patients appears indicative of AMD pathogenesis62.
Kaur et al recently reported the association of LOC387715 and HTRA1 SNPs, along with their risk estimates among Indian patients with AMD which emphasize their significant involvement in AMD susceptibility, which may be useful for predictive testing63. The role of complement in laser-induced CNV has been suggested recently64. This might explain the Janus-like context-dependent role of macrophages: the clearance of immune deposits at one time and CNV at other. The role of multigene effects in AMD is conspicuous from many published reports. Zareparsi S et al.65 have studied the susceptibility of patients with AMD who carry the variant of Toll-like receptor 4 TLR-D299G and found it to have an additive effect to AMD, in contrast to its susceptibility pattern in atherosclerosis. In contrast Edwards et al found that TLR3, TLR4, and TLR7 were unlikely to have a major impact on overall AMD risk66 and hence the deeper investigation into the role of different genes in different populations can not be ruled out. A report of the predominance of an Alu-sequence insertion in the angiotensin-converting enzyme (ACE) showed it to be protective in unaffected individuals and can be exploited as a plausible target for the dry form of AMD67. Fiotti and co-workers68 have reported a positive correlation between the existence of longer satellites (CA) in the promoter region of the gene encoding matrix metalloproteinase-9 (MMP-9) and the exudative form of AMD, prompting further analysis of complex interactions between different genes implicated in AMD. In addition, raised levels of MMP-9 in plasma revealed the association with AMD patients69. Elevated serum/plasma levels of CRP have also been associated with neovascular AMD70
Several studies have investigated the superoxidedismutase gene polymorphism in AMD and found no significant contribution towards its progression unlike other progressive disorders. The direct role of VEGF, fibroblast growth factor-2 (FGF 2) and angiopoietins in CNV has been extensively studied and established even though there are isolated reports of FGF2 gene not inhibiting CNV in the laser-injury mouse model. An intricate balance of pro- and anti-angiogenic moieties, therefore, is believed to be responsible for determining the status of CNV. For example, angiotensinogen (Ang) 1 and Ang II impart pro-angiogenic and Tie 2 (a receptor tyrosine kinase) and PEDF exert anti-angiogenic effects, inhibiting CNV. As expected, aged RPE has reduced expression of PEDF; however, it is not clear if this is due to its decreased synthesis or increased proteolytic activity in the vitreous.
Risk Factors
Genetic factors, age, cigarette smoking, nutrition, and exposure to light have been identified as AMD risk factors. According to the prevalence estimates from World Health Organization AMD is the most common cause of blindness in developed countries and it is estimated that 8 million people are blind worldwide or severely visually impaired as a result of disease. The prevalence of AMD, already high in developed countries, is likely to increase dramatically according to some estimates by 50% with aging of the population71. Smoking has been identified as one of the few modifiable risk factors for AMD, a leading cause of vision loss in older Americans. Smoking may contribute to AMD through several pathways, including reduction of antioxidant levels, decreasing blood flow around the eye or affecting the pigments (coloration) in the retina72. AMD doubles the risk of dying from a heart attack or stroke, reveals research published in the British Journal of Ophthalmology73. In the normal retina, RPE performs the functions of macrophages and a neuroprotective cell layer. During aging and with the progression of the disease, the degree of each of these functions diminishes. As a result of a complex series of molecular events, including environmental, biochemical and genetic factors, AMD develops. It is chiefly accompanied by aberrant regulation of the CFH gene (complement factor H) in cells and complement 5 (C5), immunoglobulin G (IgG), tissue inhibitor of metallo-proteinase-3 (TIMP-3) and lipofuscin deposit formation beneath the RPE, which facilitates the induction of neovascularization and the consequent blurring of vision. With the recent postulation of an association of the CFH gene with AMD and characterization of chemokine-deficient knockout mice with features resembling AMD3, considerable excitement has been generated in this field. Recent study among AMD patients and controls has revealed elevated plasma concentrations of activation products C3d, Ba, C3a, C5a, SC5b-9, substrate proteins C3, C4, regulators factor Η thus providing evidence for an association of systemic activation of the alternative complement pathway with genetic variants of CFH that were previously linked to AMD susceptibility74. Surprisingly, smoking intensity had reported to have a negative impact on general-, near-, and far visual functional status independent of AMD-status. Quitting smoking seemed to have a time-dependent protective effect on near and far vision75.
Research to understand the dynamics of RPE biology during aging has stimulated interest in preventive and prophylactic therapies for earlier intervention during the degenerative processes. At present, laser photocoagulation and/or photo-dynamic therapy (PDT) with steroids is known to slow the progression of the disease temporarily and is the only treatment available, but with better understanding of these molecular mechanisms, advanced therapeutic approaches can bring hope to patients with AMD.
Animal Models
Progress in the field of AMD has been slow because of a lack of animal models. The recent development of animal models for AMD has allowed researchers to investigate more translational and innovative therapies and pathogenetic studies of AMD76,77. Many animal models have been used to simulate AMD through high-fat diets and phototoxicity78,79, senescence acceleration80 or candidate-gene manipulation81,83, resulting in partial clinical, histological and angiographic features of the human condition. In mice fed with high-fat diets, Dithmar and coworkers reported age-dependent higher electron-lucent debris and cholesterol levels compared with those fed normal diets. There are some similarities with basal linear deposits, although the debris does not form a discrete layer external to the basement membrane of the RPE, which occurs in basal linear deposits. This animal model of Blam-D-like material in Bruch’s membrane is similar to the deposits that occur in AMD but many other clinical features, such as drusen, immune deposits and photoreceptor atrophy, are lacking. Some eyes in this older group also develop endothelial invasion into Bruch’s membrane but none of these has classic linear deposits. Cousins and co-workers79have demonstrated that age increases the capacity of dietary fat, especially in the presence of environmental light, to induce sub-RPE deposits, but these studies provide little information about the understanding of pathophysiology of AMD. Majji ef al.80 have described senescence-accelerated mice (SAMP8) as models that show choriocapillary atrophy, abnormal deposits in Bruch’s membrane and intra-Bruch’s membrane CNV, although this model doesn’t capture most of the features of AMD. Nevertheless, given the rare occurrence of CNV in mice, the study of angiogenic processes will provide insights into the secondary effects of CNV on photoreceptor disruption, which ultimately leads to deficits in visual perception. The dystrophic RCS (Royal College of Surgeons) rat is another model that undergoes progressive photoreceptor degeneration due to a primary defect in RPE cells. Although it does not present with spontaneous CNV, it is useful for assessing the responsiveness of RPE transplantation and stem-cell implantation studies84,85.
The use of VEGF-overexpressing mice under the control of the RPE65 promoter is a potential tool for analysing intrachoroidal neovascularization86. This model might be useful to screen for inhibitors of choroidal vessel growth. The currently used laser-injury model for studying CNV, postulated by Ryan et al. is another model that recapitulates some of the cardinal features of CNV to which many drug development studies have been adapted87. There is a small fraction of belly spot and tail heterozygote knockout (Bst+/-) mice that spontaneously develop CNV. However, the majority of these heterozygous mice exhibit other severe ocular disturbances88 and therefore their application for AMD might be limited. Nearly 60% of Bst+/-; mice have abnormal pupillary light responses owing to unilateral or bilateral atrophy of the optic nerves. The ocular phenotype of patients with early or late AMD bears no resemblance to the panoply of structural abnormalities in Bst+/- mice, which probably result from global embryonic dysfunction rather than the specific perturbation of cellular pathways. It is known that TIMP-3 has an important role in CNV. Sorsby fundus dystrophy (SFD) is a rare late-onset macular dystrophy caused by mutations in the TIMP3 gene. Weber and co-workers89 generated knock-in mice displaying early features of age-related changes in Bruch’s membrane and the RPE. These are good for studying the role of metalloproteinases in the pathogenesis of neovascularization. These mice, however, take a long time to display abnormalities in the inner aspect of Bruch’s membrane and the organization of the adjacent basal microvilli of the RPE. Another model for is the best vitelliform macular dystrophy (VMD) model in rats and has been recently proposed for studying AMD because it shares common features with AMD90. Bestrophin, which is a 68-kDa basolateral plasma-membrane protein that is expressed in RPE cells, is a regulatory component of Ca2+ channels and is encoded by the VMD2 gene, which is mutated in Best macular dystrophy. This model is characterized by deposits of lipofuscin in Bruch’s membrane with accompanying photoreceptor atrophy and can be used for studying the RPE function by electrooculogram. Although there are several reports of immune-mediated processes leading to drusen formation91-93, Ambati and Anand1 described the first animal model for the development of drusen and lipofuscin deposits in the RPE or spontaneously occurring CNV resembling that seen in patients with AMD. These features develop as the Cc/2-and Ccr2-knockout mice age. Ccl2 is an 8-10-kDa chemotactic cytokine that recruits monocytes to inflammatory sites. It acts through a transmembrane-domain G-protein-coupled receptor called Ccr294. RPE cells secrete Ccl-2 in a polarized fashion through their basal surface95. Through in vitro and in situ studies, Ambati and coworkers demonstrated that choroidal macrophages migrate by Ccl-2 gradient, adhere to and degrade the immune deposits. Ccl-2- and Ccr-2-deficient mice have an impaired recruitment of macrophages. It has been suggested that, as a result of this dysfunction, there is an accumulation of complement fragments that might damage RPE and induce VEGF production by these cells, resulting in the development of CNV. Although the possibility of macrophage recruitment that is independent of the Ccl-2-Ccr2 axis has not been excluded, such as through RPE trophic or anti-angiogenic factors, these eyes exhibit the morphological, ultrastructural and functional features that are characteristic of human AMD. The occurrence of CNV is subdued (in ~25% of Ccl2- and Ccr2 knockout mice) but the detection of C5, IgG, TIMP-3, SAP, advanced-glycation end products (AGEs), A2E (a component of lipofuscin) and VEGF by immunostaining and western blotting is in reasonable consonance with the features of AMD pathology. These pathologies are absent in age-matched wild-type mice and several other knockout strains of mice that were analysed, such as macrophage inflammatory protein (MIP 1α-/-)-, Β5 integrin (B5-/-)- and Chemokine receptor 5 (Ccr5-/-)-knockout mice. Even in a study of apolipoprotein Ε {apoE-/-mice), BlinD and BlanD were reported to be elevated but no drusen or CNV were observed 96. Bora and co workers97 have recently recapitulated some elements of the Ccl-2 and Ccr-2 model by reporting a reduction in laser-induced CNV in cobra-venom-depleted complement and C3-/- mice, lending support to the role of complement participation in CNV and AMD. Combined with the advancement of our understanding of a causal link between the gene encoding complement factor Η and the pathology of AMD, it has become possible to organize the inflammatory paradigm in AMD into a more cogent concept. With such developments, there is likely to be a major shift in our approach to the treatment of this disease. Recent study investigated Ccl2-/-/Cx3cr1-/- [double-knockout (DKO)] mice developed AMD-like retinal lesions such as abnormal retinal pigment epithelium cells, drusen, photoreceptor atrophyand choroidal neovascularization, which progressed with age and reversed with high omega-3 long-chain polyunsaturated fatty acid diet. A broad spectrum of AMD pathologies with early onset and high penetrance in these mice implicate certain chemokines, A2E and endoplasmic reticulum proteins in AMD pathogenesis98.
Current therapeutic approaches - changing concepts
Current therapies either target RPE or CNV. The wet, or exudative, form of macular degeneration affects 15% of those diagnosed with AMD, but accounts for ~80% of the visual complications leading to blindness. Conventional laser-photocoagulation surgery to remove CNV is indicated in only 10–20% of patients with wet AMD. After laser surgery, patients report diminished visual acuity. Likewise, PDT, which is used to halt the growth of blood vessels into the central retina, is effective in a small proportion of patients with wet AMD. Tiwari et al have reported that the visual outcomes of photodynamic therapy (PDT) with verteporfin and transpupillary thermotherapy (TTT) for classic subfoveal choroidal neovascularization (CNVM) secondary to age-related macular degeneration (ARMD) in one clinical trial. They observed the short-term preservation of vision in patients of classic CNVM due to ARMD, PDT seems to be better than TTT if the pre-laser best corrected visual acuity is > 20/63 but both are equally effective if pre-laser best corrected visual acuity is < 20/63 99. Clinicians observe that abnormal bloodvessel growth reoccurs after either laser or PDT100,101. Clearly additional therapeutic options are needed to address this problem and to restore the lost function. Considering the known effects of oxidative stress on RPE cell health and function, an obvious therapeutic approach for the maintenance of RPE cell function is nutritional supplementation with antioxidants. The value of antioxidant supplements in slowing the progression of age-related degenerative changes has recently been supported by long-term studies such as the Age-Related Eye Disease Study (AREDS)102,103. Clinical studies of dietary supplements that contain carotenoids, anti-oxidant vitamins A, C and Ε and minerals such as zinc show a 25% decrease in the rate of progression to aggressive AMD among high-risk patients. Similar in scope to vitamin supplementation and subthreshold laser, these therapies have the potential for broad use because they can be effective as preventive or prophylactic treatments.
Considerable progress has been made with experimental cellular and gene therapies for macular degeneration. The ultimate goal of experimental RPE cell transplantation is to supplement the RPE cell layer with healthy native or genetically engineered cells to prevent further atrophy of the RPE and retina. Transplantation has been performed in animal models and in limited human clinical trials; however, the desired restoration of normal function has not been conclusively demonstrated104-106. Recently, Radtke et al. demonstrated the efficacy and safety of the implantation of neural retinal progenitor cell layers in AMD patients, out of which 70% patients exhibited improved visual acuity. Thus this outcome provides clinical evidence of the safety and beneficial effect of retinal implants and corroborates results in animal models of retinal degeneration. Gene therapy to modify the expression of proteins in the RPE cell layer has the potential to prevent macular degeneration at the molecular level. Delivery of therapeutic proteins, such as antiangiogenic proteins, to the eye is a demonstrated method for the control of age-related macular degeneration (AMD). However, one of the key limitations is the requirement for frequent and repeated intraocular injections. Mc Vey et al demonstrate that repeated protein production in the eye can be stimulated from the cytomegalovirus (CMV) promoter without repeat intraocular injections using a small molecule107, all-trans retinoic acid (ATRA), which is by systemic delivery can stimulate protein production multiple times in the eye and resulted in stimulation of gene expression to relevant levels that block abnormal blood vessel growth in an experimental animal model for AMD. These data support the principles of this technological discovery to therapeutic applications for chronic ocular diseases. Strategies for treating AMD with gene transfer are being developed to address the dilemmas of systemic toxicity, immune reaction and nonspecific cellular expression. Disease-causing mutations in genes (e.g. CFH, ATP-binding cassette receptor (ABCR), TIMP3 and Ccl2 and Ccr2) that regulate interactions between the RPE and retina in two of the early-onset forms of macular degeneration might be potential targets for gene therapy108,109. An improved understanding of the role of vascular endothelial growth factor (VEGF) in the genesis of choroidal neovascular membranes has led to the creation and use of intravitreous anti-VEGF antibodies (bevacizumab and ranubizumab) and an aptamer (pegaptanib) in the treatment of these lesions. These new intravitreous injections for AMD have supplanted previous treatments in both efficacy and safety and are now the standard of care for neovascular AMD. Many clinical trials have adopted an anti-VEGF approach as a standard target for AMD. These aim to curtail the abnormal blood-vessel formation that obliterates vision. A synthetic VEGF-aptamer RNA binds to extracellular VEGF and tends to inhibit the CNV. Even soluble fms-like tyrosine kinase (sflt-1) and angiostatin delivery has been attempted in an effort to contain the CNV110. The clinically relevant inhibition of the protease system (uPA-uPAR) might prove to be a potentially novel antiangiogenic therapy for CNV if a recent report in a laser CNV model is to be believed111. Intercellular adhesion molecule (ICAM-1) blockade has also been attempted with the purpose of inhibiting the inflammatory reactions at the time of AMD112,113. Even thalidomide has been tested for its anti-angiogenic effects, but was not promising because of strong side effects114,115. Genistain has also been studied to inhibit choriocapillaries regeneration116. Perhaps there is a need to shift the focus from a destructive (CNV resolution) to a restorative paradigm. The new treatment strategy of reconstruction of the altered subretinal structure by maculoplasty (macular reconstruction) might be helpful. This serves to replenish a normal photoreceptor–RPE interface. Some groups have been attempting the use of bone-marrow-derived CD34+ cells for regeneration of the retina in experimental animal models. It remains to be seen how beneficial the effects are going to be. So far, no clear consensus of appropriate genetic targets for either dry or wet AMD has been elucidated. The restoration of visual function is a central component for the development of a clinically acceptable cell or gene therapy for AMD. The potential for a rescue-of-function effect of subretinal viral delivery of the Ccl2 and Ccr2 or CFH genes coupled with stem-cell therapy might be the rationale replete with the greater hope. It is uncertain whether this would revive the appropriate number of specialized macrophages at the site of drusen deposits, to participate in drusen degradation without relieving the inhibition of the CFH complement pathway in RPE. Although, recently the implantation of neural retinal progenitor cell layers (sheets) with its retinal pigment epithelium (RPE) in dry AMD showed improved visual acuity scores in 70% patients. This outcome provides clinical evidence of the safety and beneficial effect of retinal implants31.Treatment options for wet AMD are still at a relatively early stage of evaluation and at present no treatment for dry AMD is routinely available.
Paradoxes
In spite of rapid strides in this field, the spectrum of genetic targets involved in the pathogenesis of AMD has become more complex. Determining an appropriate target in this disease is the biggest obstacle. Because most of the information is derived from animal experiments, the absence of macula in the experimental mice is a glaring paradox in the study of AMD. However, this can be explained by the common observation that there are other areas, independent of the macula, that are also affected in AMD. Also, the prospect of using a TIMP-3-, CFH-or Ccl-2- and Ccr-2-driven restorative response in such models can provide useful information for designing future therapeutic strategies. In Ccl2-/-
Conclusion
With the rapid advances in the cell biology of AMD, it is desirable to screen plausible targets for the development of novel treatments. The reported association of CFH variants in humans and the Ccl2 and Ccr2 genes in mice has unified the inflammatory theory of AMD. Our knowledge of a role for complement and macrophages in the pathogenesis of AMD has thus become pronounced. Therefore, there is the need for a paradigm shift in the treatment approach. This could be done by developing a means for RPE reconstruction rather than the destruction of CNV by PDT, photocoagulation or therotherapy. For exploring methods to restore RPE, mechanisms of drusen resolution and the regeneration of healthy photoreceptors must be understood. Animal models can provide invaluable platforms for validating therapies, including tissue engineering and stem-cell transplantation. Ccl-2 and Ccr-2 subretinal vector-delivery experiments are ongoing for studying their rescue-of-function effect and similar experiments with CFH could be envisaged. The mode of targeting engineered stem cells is an approach that might revive the functional RPE-photoreceptor interface. Such approaches could include anti-complement and anti-inflammatory concepts. Furthermore, similar disorders, such as those characterized by atherosclerotic and senile plaque development, macrophage biology and immune-mediated mechanisms, can also be productively addressed by the progress in the field of AMD.
Competing interests - None
Received Date : 1 April 2009
Revised Date : 25 April 2009
Accepted Date : 2 May 2009
References
1. Ambati J, Anand A, Fernandez S, et al. animal model of age-related macular degeneration in senescent Ccl-2- or Ccr-2-deficient mice. Nature Medicine. 2003; 9:1390-1397.
2. Mullins R F, Russell S R, Anderson D H, et al. Drusen associated with aging and age-related macular degeneration contain proteins common to extracellular deposits associated with atherosclerosis, elastosis, amyloidosis, and dense deposit disease. The FASEB Journal. 2000; 14:835-846.
3. Chowdhury Η R, Patel Ν and Sivaprasad S. Ocular neovascularization: potential for the angiopoietin/rie-2 pathway. Expert Review of Ophthalmology 2009; 4:65-78.
4. Geniez Μ S and D’Amore PA. Development and pathology of the hyaloid, choroidal and retinal vasculature. Int. J. Dev. Biol. 2004; 48:1045-1058.
5. Zhao L, Wang Z, Liu Y, et al. Translocation of the retinal pigment epithelium and formation of sub-retinal pigment epithelium deposit induced by subretinal deposit. Molecular Vision 2007; 13:873-880.
6. Hageman G S, Luthert Ρ J, Chong Ν Η, et al. An Integrated Hypothesis That Considers Drusen as Biomarkers of Immune-Mediated Processes at the RPE-Bruch’s Membrane Interface in Aging and Age-Related Macular Degeneration. Prog Retin Eye Res 2001; 6:705-32
7. Forrester J V, Macrophages eyed in macular degeneration. Nature Medicine. 2003; 9:1350-1351.
8. Patel Μ and Chan C C. Immunopatho-logical aspects of age-related macular degeneration. Semin Immunopathol 2008; 2:97-110.
9. Lois N, Owens S L, Coco R, et al.Fundus autofluorescence in patients with age-related macular degeneration and high risk of visual loss. American Journal of Ophthalmology 2002; 3:341-349.
10. Kaarniranta Κ and Salminen A . Age-related macular degeneration: activation of innate immunity system via pattern recognition receptors. Journal of Molecular Medicine 2009; 87:117-123.
11. Xu H, Chen Μ and Forrester J V. Para- inflammation in the aging retina. Progress in Retinal and Eye Research 2009; 5:348-368.
12. Forrester J V, Lumsden L, Duncan L, et al. Choroidal dendritic cells require activation to present antigen and resident choroidal macrophages potentiate this response. British Journal of Ophthalmology 2005; 89:369-377.
13. Zahs K R, Wu T. Confocal microscopic study of glial-vascular relationships in the retinas of pigmented rats. J Comp Neurol 2001; 429:253-256.
14. Karwatowski W S, Jeffries Τ Ε, Duance V C, et al. Preparation of Bruch’s membrane and analysis of the age-related changes in the structural collagens. British Journal of Ophthalmology 1995; 79: 944-952.
15. Burke J M, CaoF, Irving Ρ Ε, et al. Expression of E-Cadherin by Human Retinal Pigment Epithelium: Delayed Expression In Vitro. Investigative Ophthalmology and Visual Science 1999; 40:2963-2970.
16. Quinn R Η and Miller SS, Ion transport mechanisms in native human retinal pigment epithelium. Investigative Ophthalmology & Visual Science 1992; 33:3513-3527.
17. Wolfensberger, T.J. The role of carbonic anhydrase inhibitors in the management of macular edema. Doc. Ophthalmol 1999; 97:387-397.
18. Versaux-Botteri C, Gibert J M, Nguyen- Legros J, et al. Molecular identification of a dopamine D1b receptor in bovine retinal pigment epithelium. Neuroscience letters 1997; 237:9-12.
19. Rendleman C R and Glickman R D. Possible therapy for age-related macular degeneration using human telomerase. Brain Research Bulletin 2004; 62:549-553.
20. Kamei Μ and. Hollyfield J G. TIMP-3 in Bruch’s Membrane: Changes during Aging and in Age-Related Macular Degeneration. Investigative Ophthalmology and Visual Science 1999; 40:2367-2375.
21. Penfold Ρ L, Madigan Μ C, Gillies Μ C et al. Immunological and aetiological aspects of macular degeneration. Prog Retin Eye Res 2001; 20 :385-414.
22. Duvall J and Tso MO. Cellular mechanisms of resolution of drusen after laser coagulation. An experimental study. Archives of ophthalmology 1985; 103:694-703.
23. Grossniklaus Η Ε, Ling J X, Wallace T M et al. Macrophage and retinal pigment epithelium expression of angiogenic cytokines in choroidal neovascularization. Molecular Vision 2002; 8:119-126.
24. Espinosa-Heidmann D G, Suner I J, Hernandez Ε Ρ, et al. Macrophage Depletion Diminishes Lesion Size and Severity in Experimental Choroidal Neovascularization. Investigative Ophthalmology and Visual Science 2003; 44:3586-3592.
25. Sakurai E, Anand A,. Ambati Β Κ, et al. Macrophage Depletion Inhibits Experimental Choroidal Neovascularization. Investigative Ophthalmology and Visual Science 2003; 44:3578-3585.
26. Ogata N, Wang L, Jo N, et al. Pigment epithelium derived factor as a neuroprotective agent against ischemic retinal injury. Curr Eye Res 2001; 22:245-52.
27. Alberdi E, Aymerich Μ S, and Becerra S P. Binding of Pigment Epithelium-derived Factor (PEDF) to Retinoblastoma Cells and Cerebellar Granule Neurons. J Biol Chem 1999; 44:31605-31612.
28. Wada M, Gelfman CM, Matsunaga H, et al. Density-dependent expression of FGF-2 in response to oxidative stress in RPE cells in vitro. Curr. Eye Res 2001; 23:226-231.
29. Henry J. Kaplan, Marc A. Leibole, Tongalp Tezel, et al. Fas ligand (CD95 ligand) controls angiogenesis beneath the retina. Nature Med 1999; 5:292-297.
30. Lambooij A C, Kliffen M, Mooy C M. et al. Role of Fas-ligand in agerelated maculopathy not established. Am. J. Ophthalmol 2001; 132:437-439.
31. Radtke Ν D, Aramant R B, Petry Η. Μ, et al. Vision Improvement in Retinal Degeneration Patients by Implantation of Retina Together with Retinal Pigment Epithelium. American Journal of Ophthalmology 2008; 2:172-182.
32. Sarna T. Properties and function of the ocular melanin:a photobiophysical view. J. Photochem. Photobiol 1992; 12:215-258.
33. Sundelin S P, Nilsson S E, Brunk U T. Lipofuscin-formation in cultured retinal pigment epithelial cells is related to their melanin content. Free Radic. Biol. Med 2001; 30:74-81.
34. Rrohan RM, Fernandez A, Udagawa T et al. Genetic heterogeneity of angiogenesis in mice. FASEBJ 2000; 14:871-876.
35. Braun M, KageA, Heimann K, et al. Retinal pigment epithelial cells from Royal College of Surgeons dystrophic rats can take up melanin granules. Graefes Arch. Clin. Exp. Ophthalmol 1999; 237:67-71.
36. Wallow I H. Repair of the pigment epithelial barrier following photocoagulation. Arch. Ophthalmol 1984; 102:126-135.
37. Eldred G E. Lipofuscin and other lysosomal storage deposits in the retinal pigment epithelium. Function and Disease. Oxford University Press 1998; 651-668.
38. Finnemann S C, Leung L W and Boulan Ε R. The lipofuscin component A2E selectively inhibits phagolysosomal degradation of photoreceptor phospholipids by the retinal pigment epithelium. Proc. Natl. Acad. Sci. U. S. A. 2002; 99:3842-3847.
39. Clark A F, Mechanism of Action of the Angiostatic Cortisene Anecortave Acetate. Survey of Ophthalmology 2007; 52:S26-S34.
40. Scholl Η Ρ Ν, Issa Ρ C. Walier Μ, et al. Systemic Complement Activation in Age-Related Macular Degeneration. PLoS ONE 2008; 3(7):e2593.
41. Francis Ρ J, Schultz D W, Hamon S, et al. Haplotypes in the Complement Factor Η (CFH) Gene: Associations with Drusen and Advanced Age-Related Macular Degeneration. PLoS ONE 2007; 2:e1197.
42. Gold B, Merriam J E, Zernant J, et al. Variation in factor Β (BF) and complement component 2 (C2) genes is associated with age-related macular degeneration. Nature Genetics 2006; 38:458-462.
43. McKay G J, Silvestri G, Patterson C C, et al. Further Assessment of the Complement Component 2 and Factor Β Region Associated with Age-Related Macular Degeneration. Investigative Ophthalmology and Visual Science 2009; 50:533-539
44. Xu Y, Guan N, Xu J, et al. Association of CFH, LOC387715, and HTRA1 polymorphisms with exudative age-related macular degeneration in a northern Chinese population. Molecular Vision 2008; 14:1373-1381.
45. Allikmets R, Shroyer NF, Singh N, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 1997; 277:1805-7.
46. Klaver CC, Kliffen M, van Duijn CM, et al. Genetic association of apolipo-protein Ε with age-related macular degeneration. Am J Hum Genet 1998; 63:200-6.
47. Stone EM, Braun TA, Russell SR, et al. Missense variations in the fibulin 5 gene and age-related macular degeneration. Ν Engl J Med 2004; 351:346-53.
48. Smith W, Assink J, Klein R, Mitchell et al. Risk factors for age-related macular degeneration: Pooled findings from three continents. Ophthalmology 2001; 108:697-704.
49. Tomany SC, Wang JJ, Van Leeuwen R, et al. Risk factors for incident age-related macular degeneration: pooled findings from 3 continents. Ophthalmology 2004; 111:1280-7.
50. Chakravarthy U, Augood C, Bentham GC, et al. Cigarette smoking and age-related macular degeneration in the EUREYE Study. Ophthalmology 2007; 114:1157-63.
51. Klein Μ L, Schultz D W, Edwards A, et al. Age-related macular degeneration. Clinical features in a large family and linkage to chromosome 1q. Arch Ophthalmol 1998; 116:1082-8.
52. Kenealy S J, Schmidt S, Agarwal A, et al. Linkage analysis for age-related macular degeneration supports a gene on chromosome 10q26. Mol Vis 2004; 10:57-61.
53. Fisher S A, Abecasis G R, Yashar Β Μ, et al. Meta-analysis of genome scans of age-related macular degeneration. Hum Mol Genet 2005; 14:2257-64.
54. Weeks D E, Conley Υ Ρ, Tsai Η J, et al. Age-related maculopathy: a genome-wide scan with continued evidence of susceptibility loci within the 1q31, 10q26, and 17q25 regions. Am J Hum Genet 2004; 75:174-89.
55. Jakobsdottir J, Conley Υ Ρ, Weeks D E, et al. Susceptibility genes for age-related maculopathy on chromosome 10q26. Am J Hum Genet 2005; 77:389-407.
56. Dean A, Liu M, Hartman S, et al. HTRA1 promoter polymorphism in wet age-related macular degeneration. Science 2006; 314:989-92.
57. Yang Z, Camp Ν J, Sun H, et al. A variant of the HTRA1 gene increases susceptibility to age-related macular degeneration. Science 2006; 314:992-3.
58. Chowers I, Cohen Y, Cohen Ν G, et al. Association of complement factor Η Y402H polymorphism with phenotype of neovascular age related macular degeneration in Israel . Molecular Vision 2008; 14:1829-1834.
59. Klein R J, Zeiss C, Chew Ε Yet et al. Complement factor Η polymorphism in age-related macular degeneration. Science 2005; 308:385-389.
60. Klaver C C W, Bakker A, DeJong P T V M, et al. Molecular genetic analysis of Ccr2 and Ccl2 in age-related macular degeneration. Invest Ophthalmol Vis Sci 2004; 45: E-Abstract3389.
61. Despriet D D G, Bergen A A B, Merriam J Ε et al. Comprehensive Analysis of the Candidate Genes CCL2, CCR2, and TLR4 in Age-Related Macular Degeneration. Investigative Ophthalmology and Visual Science 2008; 49:364-371.
62. Kaur I, Hussain A, Hussain N, et al. Analysis of CFH, TLR4, and APOE polymorphism in India suggests the Tyr402His variant of CFH to be a global marker for age-related macular degeneration. Invest Ophthalmol Vis Sci 2006; 47:3729-35.
63. Kaur I, Katta S, Hussain A, et al. Variants in the 10q26 Gene Cluster {LOC387715 and HTRA1) Exhibit Enhanced Risk of Age-Related Macular Degeneration along with CFH in Indian Patients. Investigative Ophthalmology and Visual Science 2008; 49:1771-1776.
64. Bora Ρ S, Sohn J H, Cruz J Μ C et al. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J. Immunol 2005; 174:491-497.
65. Zareparsi S, Buraczynska M, Branham KE et al. Toll-like receptor 4 variant D299G is associated with susceptibility to age-related macular generation. Hum. Mol. Genet. 2005; 14:1449-55.
66. Edwards A O, Chen D, Fridley Β L, et al. Toll-like Receptor Polymorphisms and Age-Related Macular Degeneration; Investigative Ophthalmology and Visual Science 2008; 49:1652-1659.
67. Hamdi Η Κ, Reznik J, Castellon R et al. Alu DNA polymorphism in ACE gene is protective for age-related macular degeneration. Biochem. Biophys. Res. Commun 2002; 295:668-672.
68. Fiotti N, Pedio M, Battaglia Parodi Μ et al. MMP-9 microsatellite polymorphism and susceptibility to exudative form of age-related macular degeneration. Genet. Med 2005; 7:272-277.
69. Chau Κ Υ , Sivaprasad S , Patel Ν , et al. Plasma levels of matrix metallopro-teinase-2 and -9 (MMP-2 and MMP-9) in age-related macular degeneration. Eye 2008; 22:855-859.
70. Kim I K, Ji F, Morrison Μ A et al. Comprehensive analysis of CRP, CFH Y402H and environmental risk factors on risk of neovascular age-related macular degeneration. Molecular Vision 2008; 14:1487-1495.
71. Hooper P, Jutai JW, Strong G et al. Age related Macular Degeneration and low-vision rehabilitation: a systematic review. Can j Ophthalmol 2008; 43:180-7.
72. Klein R, Knudtson Μ D; Cruickshanks Κ J, et al. Further Observations on the Association Between Smoking and the Long-term Incidence and Progression of Age-related Macular Degeneration; Arch Ophthalmol 2008; 126:115-121.
73. Tan J S L, Wang J J, Liew G, et al. Age-related macular degeneration and mortality from cardiovascular disease or stroke; British Journal of Ophthalmology 2008; 92:509-512.
74. Scholl Η Ρ Ν, Issa Ρ C, Walier Μ, et al. Systemic Complement Activation in Age-Related Macular Degeneration. PLoSONE2008; 3:e2593.
75. Neuner B, Wellmann J, Dasch B, et al. LOC387715, smoking and their prognostic impact on visual functional status in age-related macular degeneration-The Muenster Aging and Retina Study (MARS) cohort. Ophthalmic Epidemiol 2008; 15:148-54.
76. Rakoczy P, Yu M, Nusinowitz S, et al. Mouse models of age-related macular degeneration.; Exp Eye Res 2006; 86:741-752.
77. Marmorstein AD, Marmorstein LY The challenge of modeling macular degeneration in mice. Trends Genet 2007; 23:225-231.
78. Dithmar S, Sharara Ν A, Curcio C A, et al. Murine high-fat diet and laser photochemical model of basal deposits in Bruch’s membrane. Arch. Ophthalmo 2001; 119:1643-1649.
79. Cousins S.W, Espinosa-Heidmann D.G., Alexandridou Α., et al. The role of aging, high fat diet and blue light exposure in an experimental mouse model for basal laminar deposit formation. Exp. Eye Res 2002; 75:543-553.
80. Majji A B, Cao J, Chang Κ Υ, et al. Age-related retinal pigment epithelium and Bruch’s membrane degeneration in senescenceaccelerated mouse. Invest. Ophthalmol. Vis. Sci 2000; 41:3936-3942.
81. Weng J, Mata Ν L, Azarian S N, et al. Insights into the function of Rim protein in photoreceptors and etiology of Stargardt’s disease from the phenotype in ABCR knockout mice. Cell 1999; 98:13-23.
82. Dithmar S, Curcio C A, Le Ν A. et al. Ultrastructural changes in Bruch’s membrane of apolipoprotein E-deficient mice. Invest. Ophthalmol. Vis. Sci 2000; 41:2035-2042.
83. Rakoczy Ρ Ε, Zhang D, Robertson Τ et al. Progressive age-related changes similar to age-related macular degeneration in a transgenic mouse model. Am. J. Pathol 2002; 161:1515-1524.
84. Sauve Y, Girman S V, Wang S, et al. Preservation of visual responsiveness in the superior colliculus of RCS rats after retinal pigment epithelium cell transplantation. Neuroscience 2002; 114:389-401.
85. Schraermeyer U, Thumann G, Luther T, et al. Subretinally transplanted embryonic stem cells rescue photoreceptor cells from degeneration in the RCS rats. Cell Transplant 2001; 10:673-680.
86. Schwesinger C, Yee C, Rohan R M, et al. Intrachoroidal neovascularization in transgenic mice overexpressing vascular endothelial growth factor in the retinal pigment epithelium. AmJ. Pathol 2001; 158:1161-1172.
87. Pharmacological Therapy for Macular Degeneration Study Group. Interferon a-2a is ineffective for patients with choroidal neovascularization secondary to age-related macular degeneration. Results of a prospective randomized placebocontrolled clinical trial. Arch. Ophthalmol 1997; 115:865-872.
88. Smith RS, John SW, Zabeleta A, et al. The Bst locus on mouse chromosome 16 is associated with age-related subretinal neovascularization. Proc. Natl. Acad. Sci. U. S. A. 2000; 97:2191-2195.
89. Weber Β Η F, Lin B, White K. et al. A mouse model for Sorsby fundus dystrophy. Invest. Ophthalmol. Vis. Sci. 2002; 43:2732-2740.
90. Marmorstein AD, Stanton JB, Yocom J, et al. A model of best vitelliform macular dystrophy in rats. Invest. Ophthalmol. Vis. Sci. 2004; 45:3733-3739.
91. Crabb JW, Miyagi M, Gu X, et al. Drusen proteome analysis. An approach to the etiology of age-related macular degeneration. Proc. Natl. Acad. Sci. U. S. A. 2002; 99:14682-14687.
92. Hageman GS, Mullins RF, Russell SR, et al. Vitronectin is a constituent of ocular drusen and the vitronectin gene is expressed in human retinal pigmented epithelial cells. FASEB J. 1999; 13:477-484.
93. Ozaki S, Staples Μ Κ, Erickson Ρ A et al. A potential role for immune complex pathogenesis in drusen formation. Exp. Eye Res. 2000; 70:441-449.
94. Sallusto F, Mackay CR, Lanzavecchia A, et al. The role of chemokine receptors in primary, effector, and memory immune responses. Annu. Rev. Immunol. 2000; 18:593-620.
95. Holtkamp G M,De Vos A F,Peek R, et al. Analysis of the secretion pattern of monocyte chemoattractant protein-1 (MCP-1) and transformaing growth factory-β2 (TGF-β2) by human retinal pigment epithelium cells. Clin. Exp. Immunol. 1999; 118:35-40.
96. Kliffen M, Lutgens E, Daemen M, et al. The APO-/- E3-Leiden mouse as an animal model for basal laminar deposit. Br. J. Ophthalmol. 2000; 84:1415-1419.
97. Puran S B, Sohn J H, Jose Μ C et al. Role of complement and complement membrane attack complex in laser-induced choroidal neovascularization. J. Immunol. 2005; 174:491-497
98. Chan C C, Robert J R, Shen D, et al. Ccl2/Cx3cr1-Deficient Mice: An Animal Model for Age-Related Macular Degeneration. Ophthalmic Res. 2008; 40:124-128.
99. Hem Κ Τ, Prakash, Azad R. V, et al. A pilot trial for comparison of photodynamic therapy and transpupillary thermo-therapy for the management of classic subfoveal choroidal neovascularization secondary to age-related macular degeneration; Indian Journal of Ophthalmology 2007 ; 4:277-281.
100. Macular Photocoagulation Study Group. Persistent and recurrent neovascularization after laser photocoagulation for subfoveal choroidal neovascularization of agerelated macular degeneration. Arch. Ophthalmol. 1994; 112:489-499.
101. Treatment of Age-Related Macular Degeneration with Photodynamic Therapy (TAP) Study Group. Verteporfin therapy of subfoveal choroidal neovascularization in patients with age-related macular degeneration. Arch. Ophthalmol. 2002; 120:1443-1454.
102. Sackett CS, Schenning S. The age-related eye disease study: the results of the clinical trial. lnsight 2002; 27:5-7.
103. The Age-Related Eye Disease Study Research Group. A randomized, placebo-controlled clinical trial of high-dose supplementation with vitamins C and Ε, β carotene, and zinc for age-related macular degeneration and vision loss, AREDS Report. Arch. Ophthalmol. 2001; 119:1417-1436.
104. Coffey P.J. and Girman S. and Wang et al. Long term preservation of cortically dependent visual function in RCS rat by transplantation. Nat. Neurosci. 2002; 5:53-56.
105. Wang H, Leonard D S, Alessandro A Castellarin et al. Short-term study of allogeneic retinal pigment epithelium transplants onto debrided Bruch’s membrane. Invest. Ophthalmol. Vis. Sci. 2001; 42:2990-2999.
106. James M, Mark S, Eugene De Jr, et al. Allogenic fetal retinal pigment epithelial cell transplant in a patient with geographic atrophy. Retina 1999; 19:540-545.
107. McVey D, Hamilton Μ Μ, Hsu C, et al. Repeat administration of proteins to the eye with a single intraocular injection of an adenovirus vector. Mol Ther. 2008 Aug;16:1444-9.
108. Allikmets R, Shroyer Ν F, Singh N, et al. Mutation of the Stargardt disease gene (ABCR) in age-related macular degeneration. Science 1997; 277:1805-1807.
109. Felbor U, Stöhr H, Amann Τ et al. A second independent Tyr168Cys mutation in the tissue inhibitor of metalloproteinases-3 (TIMP3) in Sorsbys fundus dystrophy. J. Med. Genet. 1996; 33:233-236.
110. Lai C C, Wu W C, Chen S L. et al. Suppression of choroidal neovascularization by adeno-associated virus vector expressing angiostatin. Invest. Ophthalmol. Vis. Sci. 2001; 42:2401-2407.
111. Das, Α., Boyd N, Jones Τ R et al. Inhibition of choroidal neovascularization by a peptide inhibitor of the urokinase plasminogen activator and receptor system in a mouse model. Arch. Ophthalmol. 2004; 122:1844-1849.
112. Miyamoto K, Khosrof S, Bursell S E. et al. Vascular endothelial growth factor (VEGF)-induced retinal vascular permeability is mediated by intercellular adhesion molecule-1 (ICAM-1). Am. J.Pathol. 2000; 156:1733-1739.
113. Miyamoto K, Khosrof S, Bursell S Ε et al. Prevention of leukostasis and vascular leakage in streptozotocin-induced diabetic retinopathy via intercellular adhesion molecule-1 inhibition. Proc.Natl. Acad. Sci. U. S. A. 1999; 96:10836-10841.
114. Amato R J, Loughnan Μ S, Flynn E, et al. Thalidomide is an inhibitor of angiogenesis. Proc. Natl. Acad. Sci. U. S. A. 1994; 91:4082-4085.
115. Kruse F E, Joussen A M, Rohrschneider Κ et al. Thalidomide inhibits corneal angiogenesis induced by vascular endothelial growth factor. Graefes Arch Clin Exp Ophthalmol. 1998; 236:461-6.
116. Majji A B, Hayashi A, Kim Η C et al. Inhibition of Choriocapillaris Regeneration with Genistein. Invest Opbthalmol Vis Set.1999; 40:1477-1486.
117. Ambati J,.Ambati B, Yoo S, S.lanchulev et al. Age-Related Macular Degeneration: Etiology, Pathogenesis, and Therapeutic Strategies. Survey of Ophthalmology 2003; 48:257-293.
118. Ho AC, Maguire MG, Yoken J et al. Laser-induced drusen reduction improves visual function at 1 year. Ophthalmologyl 999; 106:1367-1373.
119. Olk R. J., Friberg T. R. .Stickney K. L. et al. Therapeutic benefits of infrared (810-nm) diode laser macular grid photocoagulation in prophylactic treatment of nonexudative age-related macular degeneration: Two-year results of a randomized pilot study. Ophthalmology 1999; 106:2082-2090.
120. Young J D, MacDonald Μ Κ, and McKechnie Ν Μ. Fundus changes in (type II) mesangiocapillary glomerulonephritis simulating drusen: a histopathological report. Br J Ophthalmol. 1989; 73:297-302.
121. Anand A, Skufca D W, Ambati Β K et al. Vascular endothelial growth factor exerts anti-inflammatory effects on human retinal pigment epithelial cells. A potential mechanism for leukocyte compartmentalization. Invest Ophthalmol Vis Sci 2003; 44:3947.
(c) Annals of Neurosciences.All Rights Reserved