Annals of Neurosciences, Vol 18, No 1 (2011)
Annals of Neurosciences, Volume 18, Issue 1 (January), 2011
Zinc Finger Nucleases: A new era for transgenic animals
ABSTRACT
The rational engineering of eukaryotic genomes would facilitate the study of heritable changes in gene expression and offer enormous potential across basic research, drug-discovery, bioproduction and therapeutic development. A significant advancement toward this objective was achieved with the advent of a novel technology that enables high-frequency and high-fidelity genome editing via the application of custom designed zinc finger nucleases (ZFNs). A ZFN is a chimeric protein that consists of the non-specific endonuclease domain of FokI fused to a DNA-binding domain composed of an engineered zinc-finger motif. Within these chimeric proteins, the DNA binding specificity of the zinc finger protein determines the site of nuclease action.Once the engineered ZFNs recognize and bind to their specified locus, it leads to the dimerization of the two nuclease domains on the ZFNs to evoke a double-strand break (DSB) in the targeted DNA. The cell then employs the natural DNA repair processes of either non-homologous end joining (NHEJ) or homology-directed repair (HDR) to repair the targeted break. Due to the imperfect fidelity of NHEJ, a proportion of DSBs within a ZFN-treated cellular population will be misrepaired, leading to cells in which variable heterogeneous genetic insertions or deletions have been made at the target site. Alternatively, the HDR repair pathway enables precise insertion of a transgene or other defined alterations into the targeted region. By this approach, a donor template containing the transgene flanked by sequences that are homologous to the regions either side of the cleavage site is co-delivered into the cell along with the ZFNs. By creating a specific DSB, these cellular repair mechanisms are harnessed to generate precisely targeted genomic edits resulting in both cell lines and animal models with targeted gene deletions, integrations, or modifications. This review will discuss the development, mechanism of action, and applications of ZFN technology to genome engineering and the creation of animal models.
KEYWORDS : Zinc Finger Nuclease, DNA Repair, Non-homologous End Joining, Homologous Recombination, Cell Lines, Animal Models
Corresponding Author : John T. Swarthout PhD, Sigma-Aldrich Life Sciences, St. Louis, MO 63103, USA, E-mail john.swarthout@sial.com, Tel: (314) 456-7890
doi : 10.5214/ans.0972.7531.1118109
Introduction
Living organisms are composed of a convoluted network of physiological systems presenting scientists with the daunting challenge of untangling this biological web. The process of deconvolution necessitates that we ask the appropriate questions, that experiments are well conceived and controlled, and that suitable model systems are available. Animal models facilitate our understanding of human physiology, help us acquire insight into disease pathology, and enable evaluation of novel therapeutic strategies for efficacy and safety prior to initiation of human trials.
To ensure experimental success in a given animal model, it is essential to employ well-designed experiments and to use the most optimal animal models that closely mimic the human biology. This is especially critical in drug development where it is important to predict the true measure of a drug’s efficacy in order to reliably translate the results to the clinic. This is largely dependent upon the underlying biological system used for research and drug development rather than merely on the study or drug design. The challenge has been to successfully optimize the animal model that would provide construct, face and pharmacological validity.
In order to proceed in a timely, efficient, and accurate manner in our pursuit of the development of more efficacious and safer drugs, there is a real need for novel animal models to gain a more complete understanding of disease pathophysiology and to facilitate more reproducible in vivo efficacy studies. As we discuss below, the advent of the zinc finger nuclease (ZFN) technology affords researchers the ability to create novel, and translationally relevant animal models by performing targeted genetic modifications.1
Model organisms and the zinc finger nuclease technology
True insight into the complex interactions underlying biological pathways and disease pathologies requires studying these processes in the context of biological systems. In the late 1980s, homologous recombination (HR)-based gene targeting in mouse embryonic stem (mES) cell was first achieved and quickly accepted as a revolutionary methodology for genome modification. As a result, the mouse became the most popular animal model system today. The immense impact it has since made on biomedical research won the technology the 2007 Nobel Prize on Physiology or Medicine.2
While mice have proven to be a useful model and techniques have been developed for routine disruption of their genes, in many circumstances rats are considered a superior laboratory animal for studying and modeling human disease. Rats are more similar to humans physiologically and are a better model for human cardiovascular disease, diabetes, and arthritis; for autoimmune, neurological, behavioral and addiction disorders; as well as for neural regeneration, transplantation, and wound and bone healing.3,4 In addition, rat models are excellent for testing the pharmacodynamics and toxicity of potential therapeutic compounds.5 Finally, their larger size makes rats more conducive to study by instrumentation, and facilitates manipulation such as blood sampling, nerve conduction, and surgeries.
An enormous effort has been mounted to establish a similar knockout rat approach by manipulating rat ES cells. Until recently, the creation of rat models required manipulation of the genome using ionizing radiation,6 chemical-induced mutagenesis,7-10 or mobile DNA (jumping gene) technology.11,12 However, the random nature of these mutations represents a major limitation to studying gene function. Random chemical mutagenesis using the alkylating agent N-ethyl-N-nitrosourea (ENU) and mobile DNA technology using retrotransposons and transposons were the principle technologies used to generate knockout rat models and have been employed to generate mutant rat strains and potential disease models.13,14 However, mutagenesis using ENU is time-consuming and expensive, creates a high frequency of random mutations, and mapping mutations responsible for a particular phenotype is often difficult.8-10 Mobile DNA platforms permit random mutagenesis directly in the germ cells (sperm or oocytes) of mammalian model organisms, including rats, resulting in complete and stable gene disruptions at a high frequency. However, mutations are randomly disrupted throughout the entire genome and targeted genomic modulation is not possible.11,12
In 2008 rat stem cells were successfully isolated15,16 enabling the creation of a p53 knockout rat using HR.17 While this is an important achievement, the ZFN-mediated gene knockout does not require the establishment of ES cell culture, but simply requires embryo isolation, injection, and implantation using well characterized protocols. Importantly, the ZFN method suggests a general solution to targeted gene knockout in other species. This is because many animal species have embryo-manipulation methods robust enough to support the creation of transgenic animals, but do not have protocols for the establishment of ES cell culture.
The ZFN technology provides a means for efficient and targeted mutagenesis of genomic DNA in both cells and embryos. This achievement spawned from the discovery and subsequent manipulation of zinc finger protein domains which, when combined with the non-specific cleavage domain of the Fok l endonuclease, culminated in the formation of ZFNs.18 Engineered ZFNs are able to recognize and bind to a specified locus and evoke a double-stranded break (DSB) in the targeted DNA with high efficiency and base-pair precision. For targeted cleavage, two ZFN subunits (Figure 1) are designed that recognize the target sequence in a tail-to-tail conformation, with each monomer binding to half-sites that are separated by a 5-7 base pair spacer sequence.19 The targeted DSB stimulates the cells endogenous DNA repair processes of either homology-directed repair (HDR) or non-homologous end-joining (NHEJ) to heal the targeted break resulting in targeted integration or gene disruptions, respectively, at efficiencies in orders of magnitude higher than spontaneous homologous recombination.20,21
ZFN technology brings promise for more flexible and efficient creation of animal models, including species that have lacked convenient genetic tools. Successful examples are targeted knockouts in both rats22,23 and mice.24,25 Furthermore, development is well under way for the creation of knockout animal models focused on application for neurobiology, cardiovascular biology, immunology, and toxicology, as well as the creation of models harboring targeted integrations (SAGE™ Labs, St. Louis, MO).
In 2009, a collaboration involving Sigma-Aldrich, Sangamo Biosciences, the Medical College of Wisconsin, OMT Inc., and INSERM resulting in the first targeted knockout rats generated via microinjection of ZFNs into single-cell embryos.22 Guerts et al. used ZFNs to target the GFP sequence of a transgenic rat26 and successfully knocked out the gene with no off-target effects. Turning to an endogenous rat gene, the team disrupted the Immunoglobulin M gene, and both disruptions were transmitted through the germline efficiently. A similar approach was taken for the generation of a knockout rat model for SCID23 and targeted gene knockouts in mice,24,25 demonstrating the applicability of the ZFN technology.
Neurodegenerative disorders and rat neural models
Many neurodegenerative disorders like Alzheimer’s disease, Huntington’s disease, Parkinson’s disease, Schizophrenia and amyotrophic lateral sclerosis, display atypical protein assemblies or aggregates as well as induced cell death.27,28 The therapeutic avenues available today either target the sympotomatology of the disorders, or induced significant side effects to preclude prolonged and continued administration. A paucity of optimal therapeutic avenues for a majority of neurodegenerative disorders stems from our lack of understanding of the underlying pathophysiology. The current knowledge about neurodegenerative disorders has been gleaned primarily from mouse models. However, arguably the neural circuitry in rats is more similar to humans and the stability of behavioral performance in the rats allows more reliable experimentation. The larger size of rats, in particular the brain and cerebral spinal fluid (CSF) compartment, presents a technical advantage enabling availability of a larger amount of specimen for evaluation. The rats are also more amenable to surgical manipulations, allowing incorporation of newer tools like optogenetics to study the pathophysiology of neuronal disorders.13
Parkinson’s disease
Parkinson’s disease (PD) belongs to a group of conditions called movement disorders and is a neurodegenerative disorder of the central nervous system that impairs motor skills and speech.29 To date, there are no therapeutic interventions proven to halt or slow the progression of PD. The only option is the application of symptomatic therapies, such as dopamine supplementation used to offset the diminished dopaminergic output, but this type of treatment only alleviates the symptomatic motor dysfunction, not for the underlying pathology of PD.30 Therefore, it is absolutely essential to create translationally relevant animal models for the development of new therapeutic strategies targeting the actual pathogenic process.31 About 10% - 20% of patients are known to have monogenic forms of this disease32 and the SAGE labs has partnered with the Michael J. Fox Foundation to perform targeted knockout in rats of several of these genes. The targeted genes include leucine rich repeat kinase 2 (LRRK2), Parkin (PARK2), DJ-1 (PARK7), and PTEN-induced novel kinase 1 (PINK1).
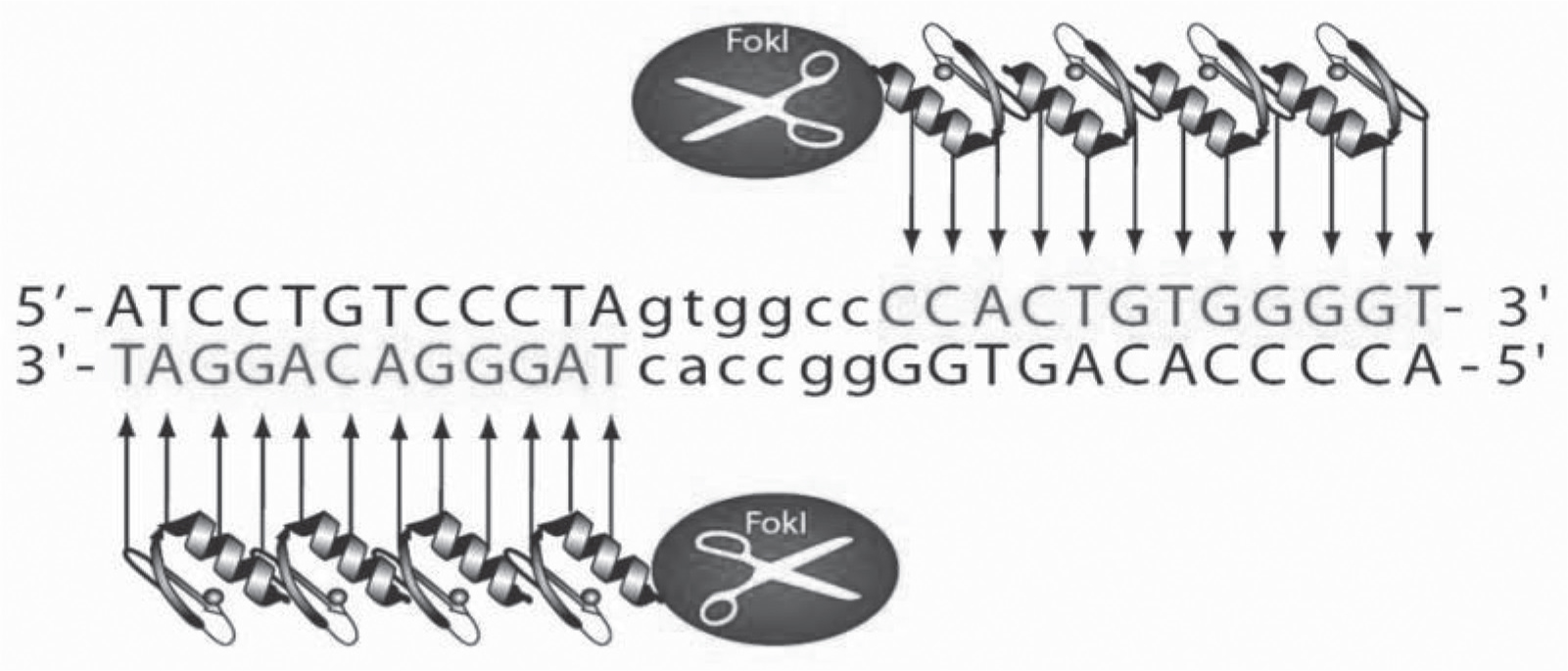
Fig. 1: Zinc Finger Nucleases are highly-specific genomic scissors. The Fok I domain must dimerize to achieve efficient DNA cleavage, requiring two ZFNs to bind at or near the cleavage site. The ZFNs only cut upon dimerization of the separate Fok I domains. By linking four zinc fingers in tandem for each of the two ZFNs, a combined recognition site of 24 base pairs is attained, specifying a unique address within the genome. These target sequences must be separated by 5-7 base pairs to allow formation of the catalytically active Fok I dimer. These positional constraints, therefore, drive a very high degree of specificity. The net result is a targeted double strand break that stimulates the cell endogenous repair machinery.
A common feature to all of these genes is that an abnormality in the structure and/or function of the expressed proteins is implicated in the pathophysiology of PD. LRRK2 encodes a protein present largely in the cytoplasm but IT also associates with the mitochondrial outer membrane and is involved in MAPKK activation, cell death, and regulation of protein ubiquitination.33,34 Autosomal-dominant LRRK mutations account for 5-6% of familial PD and 1-3% in sporadic PD, which collectively result in the most common cause of PD, making this an important model for the study of PD and apoptosis.
PARK2 localizes to the cytoplasm in neurons of many regions of the brain. It is a component of a multiprotein E3 ubiquitin ligase complex that mediates the targeting of substrate proteins for proteasomal degradation and may help degrade proteins that are toxic to neurons. Autosomal-recessive mutations in PARK2 account for at least 50% of familial PD cases with roughly 20% of patients with PD having an onset before the age of 40 years.35,36
The PINK1 protein kinase localizes to the mitochondria and is thought to protect cells from stress-induced mitochondrial dysfunction. Autosomal-recessive mutations within this gene are the second most common form of PD, contributing to almost 1% - 7% of early-onset cases.37
Finally, autosomal-recessive mutations in DJ-1 (PARK7) results in early-onset PD.38 DJ-1 is a member of the peptidase C56 family of proteins that functions as a positive regulator of androgen receptor-dependent transcription.39 Unfortunately, existing knockout mice models for PARK2, PINK1, and DJ-1 do not exhibit any major abnormality and fail to replicate the critical features of PD.31 Consequently, there is a critical need for the development of translationally relevant animal models in the rat.
Alzheimer’s disease
Alzheimer’s Disease (AD) is a neurodegenerative disorder clinically characterized by a progressive impairment in memory and cognition, and accounts for the vast majority of dementia patients.40,41 AD is characterized by the accumulation of two proteins, amyloid beta (Aβ) and tau, as well as cortical atrophy. Amyloid beta is processed from amyloid precursor protein (APP) through the sequential cleavage by β- and γ-secretase.42 Tau proteins function to stabilize microtubules and hyperphosphorylation of the protein resulting in the accumulation of intraneuronal inclusions known as neurofibrillary tangles. These two lesions are thought to contribute to the pathogenesis of AD. As with PD, the available treatment options for AD patients are directed toward reducing the symptoms and delaying the progression, but there is no cure for either of two diseases. Existing mouse models have contributed significantly to our understanding of disease pathology. However, there are some significant limitations, including differences in the biology of AD pathogenesis between humans and mice, and the limitation of behavioral flexibility and stability for learning paradigms in mice. The smaller brain and CSF volume in mice limits optimal tissue harvest and collection of CSF for evaluation of the progress of the disease, especially in the context of chronic dosing paradigms. Furthermore, the larger brain of rats enables the surgical manipulations required to integrate novel neuroscience tools like optogenetics.
SAGE Labs has pioneered several rat models to study AD which includes knockout lines for synuclein, apolipoprotein E (ApoE), and brain-derived neurotrophic factor (BDNF). Alpha-synuclein, plays a role in synaptogenesis and organization, is believed to regulate dopamine and has been implicated in several neurodegenerative diseases including both PD and AD. Mutations within the gene are also present in some Alzheimer’s disease cases, making this model important for the study neurodegeneration.
ApoE is expressed in the liver, intestines and brain where it is essential for the normal metabolism of lipids and preventing the accumulation of cholesterol-rich particles in plasma. ApoE exists in three different isoforms (E2, E3 and E4) and heterozygous and homozygous individuals for the APOE e4 allele, have approximately a 3- or 15-fold increased risk of developing AD compared with those that do not carry an e4 allele.43 Widely studied for its role in cardiovascular disease and lipoprotein transport, it has more recently been implicated in Alzheimer’s disease and cognition, making this a useful model for the study of atherosclerosis, Alzheimer’s and nerve injury.
BDNF is a neurotrophin that contributes to the growth and differentiation of neurons in the hippocampus, cortex and forebrain.44,45 Deficiency in BDNF levels has been linked to a host of neurological diseases, including AD, depression, schizophrenia and dementia, making this an important model for studying the neurodegenerative disorders like AD.46
In addition to targeted knockouts, SAGE Labs is using the ZFN technology to create rat lines over expressing APPswe/Indiana mutation, presenilin-1 mutation, and P301L tau mutation. Presenilin-1 is a component of γ-secretase which regulates APP processing. Therefore, the expression of transgenes encoding genes harboring any or these mutations will result in the formation of the characteristic lesions associated with AD.
Conclusions
ZFNs offer enormous potential across basic research, drug-discovery, bioproduction and therapeutic development, by facilitating the creation of gene knockouts and targeted integrations into more relevant backgrounds. ZFN-mediated genome editing can be used to generate novel animal models of disease that more closely mimic human disease, as well as providing more realistic data on the potential toxicity of drugs. Most importantly, the creation of knockout rats and mice using the ZFN technology does not require the establishment of ES cell culture and this fact provides a general solution for targeted gene knockout in other species. The extension of the ZFN technology for the creation of targeted gene knockouts or integrations in higher-level mammals will allow researchers to ask specific biological questions in the model systems most appropriate for their research.
The article complies with International committee of Medical Journal Editor’s uniform requirements for the manuscripts.
Competing interests – All authors are full-time employees of Sigma-Aldrich Corporation.
Received Date : 02 November 2010
Revised Date: 06 January 2011
Accepted Date : 20 January 2011
References
1. Urnov FD, Rebar EJ, Holmes MC, et al. Genome editing with engineered zinc finger nucleases. Nat Rev Genet 2010; 11: 636–646.
2. Capecchi MR. Gene targeting in mice: functional analysis of the mammalian genome for the twenty-first century. Nat Rev Genet 2005; 6: 507–512.
3. Abbott A. Laboratory animals: the Renaissance rat. Nature 2004; 428: 464–466.
4. Cozzi J, Fraichard A and Thiam K. Use of genetically modified rat models for translational medicine. Drug Discov Today 2008; 13: 488–494.
5. Lindblad-Toh, K. Genome sequencing: three’s company. Nature 2004; 428: 475–476.
6. Graf LH, Jr and Chasin LA. Direct demonstration of genetic alterations at the dihydrofolate reductase locus after gamma irradiation. Mol Cell Biol 1982; 2: 93–96.
7. O’Brien TP and Frankel WN. Moving forward with chemical mutagenesis in the mouse. J Physiol 2004; 554: 13–21.
8. Zan Y, Haag JD, Chen KS, et al. Production of knockout rats using ENU mutagenesis and a yeast-based screening assay. Nat Biotechnol 2003; 21: 645–651.
9. Smits BM, Mudde JB, van de Belt J, et al. Generation of gene knockouts and mutant models in the laboratory rat by ENU-driven target-selected mutagenesis. Pharmacogenet Genomics 2006; 16: 159–169.
10. Wienholds E, Van Eeden F, Kosters M, et al. Efficient target-selected mutagenesis in zebrafish. Genome Res 2003; 13: 2700–2707.
11. Kitada K, Ishishita S, Tosaka K, et al. Transposon-tagged mutagenesis in the rat. Nat Methods 2007; 4: 131–133.
12. Lu B, Geurts AM, Poirier C, et al. Generation of rat mutants using a coat color-tagged Sleeping Beauty transposon system. Mamm Genome 2007; 18: 338–346.
13. Bugos O, Bhide M and Zilka N. Beyond the rat models of human neurodegenerative disorders. Cell Mol Neurobiol 2009; 29: 859–869.
14. Aitman TJ, Critser JK, Cuppen E, et al. Progress and prospects in rat genetics: a community view. Nat Genet 2008; 40: 516–522.
15. Buehr M, Meek S, Blair K, et al. Capture of authentic embryonic stem cells from rat blastocysts. Cell 2008; 135: 1287–1298.
16. Li P, Tong C, Mehrian-Shai R, et al. Germline competent embryonic stem cells derived from rat blastocysts. Cell 2008; 135: 1299–1310.
17. Tong C, Li P, Wu NL, et al. Production of p53 gene knockout rats by homologous recombination in embryonic stem cells. Nature 2010; 467: 211–213.
18. Kim YG, Cha J and Chandrasegaran S. Hybrid restriction enzymes: zinc finger fusions to Fok I cleavage domain. Proc Natl Acad Sci U S A 1996; 93: 1156–1160.
19. Bibikova M, Carroll D, Segal DJ, et al. Stimulation of homologous recombination through targeted cleavage by chimeric nucleases. Mol Cell Biol 2001; 21: 289–297.
20. Durai S, Mani M, Kandavelou K, et al. Zinc finger nucleases: custom-designed molecular scissors for genome engineering of plant and mammalian cells. Nucleic Acids Res 2005; 33: 5978–5990.
21. Porteus MH and Carroll D. Gene targeting using zinc finger nucleases. Nat Biotechnol 2005; 23: 967–973.
22. Geurts AM, Cost GJ, Freyvert Y, et al. Knockout rats via embryo microinjection of zinc-finger nucleases. Science 2009; 325: 433.
23. Mashimo T, Takizawa A, Voigt B, et al. Generation of knockout rats with X-linked severe combined immunodeficiency (X-SCID) using zinc-finger nucleases. PLoS One 2010; 5: 8870.
24. Carbery ID, Ji D, Harrington A, et al. Targeted Genome Modification in Mice Using Zinc Finger Nucleases. Genetics 2010; 186: 451–459.
25. Meyer M, De Angelis MH, Wurst W, et al. Gene targeting by homologous recombination in mouse zygotes mediated by zinc-finger nucleases. Proc Natl Acad Sci USA 2010; 107: 15022–15026.
26. Michalkiewicz M, Michalkiewicz T, Geurts AM, et al. Efficient transgenic rat production by a lentiviral vector. Am J Physiol Heart Circ Physiol 2007; 293: 881–894 .
27. Bredesen DE, Rao RV and Mehlen P. Cell death in the nervous system. Nature 2006; 443: 796–802.
28. Rubinsztein DC. The roles of intracellular protein-degradation pathways in neurodegeneration. Nature 2006; 443: 780–786.
29. Jankovic J. Parkinson’s disease: clinical features and diagnosis. J Neurol Neurosurg Psychiatry 2008; 79: 368–376.
30. Savitt JM, Dawson, VL and Dawson TM. Diagnosis and treatment of Parkinson disease: molecules to medicine. J Clin Invest 2006; 116: 1744–1754.
31. Dawson TM, Ko HS and Dawson VL. Genetic animal models of Parkinson’s disease. Neuron 2010; 66: 646–661.
32. Lesage S and Brice A. Parkinson’s disease: from monogenic forms to genetic susceptibility factors. Hum Mol Genet 2009; 18: R48–59.
33. Paisan-Ruiz C, Jain S, Evans EW, et al. Cloning of the gene containing mutations that cause PARK8-linked Parkinson’s disease. Neuron 2004; 44: 595–600.
34. Smith WW, Pei Z, Jiang H, et al. Leucine-rich repeat kinase 2 (LRRK2) interacts with parkin, and mutant LRRK2 induces neuronal degeneration. Proc Natl Acad Sci U S A 2005; 102: 18676–18681.
35. Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 1998; 392: 605–608.
36. Lucking CB, Durr A, Bonifati V, et al. Association between early-onset Parkinson’s disease and mutations in the parkin gene. N Engl J Med 2000; 342: 1560–1567.
37. Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Rev Mol Med 2009; 11: e22.
38. Bonifati V, Rizzu P, van Baren MJ, et al. Mutations in the DJ-1 gene associated with autosomal recessive early-onset parkinsonism. Science 2003; 299: 256–259.
39. Niki T, Takahashi-Niki K, Taira T, et al. DJBP: a novel DJ-1-binding protein, negatively regulates the androgen receptor by recruiting histone deacetylase complex, and DJ-1 antagonizes this inhibition by abrogation of this complex. Mol Cancer Res 2003; 1: 247–261.
40. Ferri CP, Prince M, Brayne C, et al. Global prevalence of dementia: a Delphi consensus study. Lancet 2005; 366: 2112–2117.
41. Sleegers K, Lambert JC, Bertram L, et al. The pursuit of susceptibility genes for Alzheimer’s disease: progress and prospects. Trends Genet 2010; 26: 84–93.
42. Brouwers N, Sleegers K and Van Broeckhoven C. Molecular genetics of Alzheimer’s disease: an update. Ann Med 2008; 40: 562–583.
43. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 1993; 261: 921–923.
44. Barde YA, Edgar D and Thoenen H. Purification of a new neurotrophic factor from mammalian brain. EMBO J 1982; 1: 549–553.
45. Binder DK and Scharfman HE. Brain-derived neurotrophic factor. Growth Factors 2004; 22: 123–131.
46. Murer MG, Yan Q and Raisman-Vozari R. Brain-derived neurotrophic factor in the control human brain, and in Alzheimer’s disease and Parkinson’s disease. Prog Neurobiol 2001; 63: 71–124.
(c) Annals of Neurosciences.All Rights Reserved