Annals of Neurosciences, Vol 16, No 1 (2009)
Annals of Neurosciences, Volume 16, Issue 1 (January), 2009
Polycyclic aromatic hydrocarbons in air and their neurotoxic potency
in association with oxidative stress : A brief perspective
ABSTRACT
Polycyclic aromatic hydrocarbons (PAHs) are a class of toxic organic chemicals widely distributed in the environment and in food stuffs. PAHs such as Benzo a pyrene, [B (a) P] essentially enters either through the ingestion of contaminated food and water or by the inhalation of particulates in the ambient air. The link between B (a) P metabolism and oxidative damage appears to be one of the key pleiotropic modulators which may be involved with several pathological processes because of its high affinity for lipid-rich tissues such as brain. B (a) P can enhance the generation of reactive oxygen species (ROS) by inducing cytochrome P450 enzymes and free radicals produced by B (a) P metabolism. This can alter physiological functions like neuronal development, differentiation, and signal transduction. Brain does not have a strong antioxidant defense system and has limited or poor ability to replace adult neurons. The molecular mechanism of oxygen derived species such as superoxide radicals, hydrogen peroxide, singlet oxygen and hydroxyl radicals produced by B (a) P metabolism and its implication in the etiology of wide array of neurological disorders remain elusive. ROS can be involved in the neuropathology of bipolar disorder, schizophrenia, and possible reduction in cognitive abilities among infants, and it is known to play janus like role of possessing both deleterious and beneficial effects. This review discusses the role of oxidative stress derived from B (a) P metabolism relating to neurological disorders, which can introduce new targets for the development of therapeutic interventions.
KEY WORDS : Polycyclic Aromatic Hydrocarbons, PAH; Benzo (a) pyrene , Reactive Oxygen Species (ROS), Oxidative stress. Behavioral Neurotoxicity
Corresponding Author: Prof. P. Prakash Babu, E-mail : ppbsl@uohyd.ernet.in
doi: 10.5214/ans.0972.7531.2009.160109
Introduction
Polycyclic aromatic hydrocarbons (PAHs) are ubiquitous environmental pollutants, among which benzo (a) pyrene (B (a) P) is a PAH having five aromatic rings in a fused, honey comb-like structure. B (a) P is toxic to humans and laboratory animals and is generated through the burning of fossil fuels or wood, and is notably found in diesel exhaust particles, cigarette smoke, charcoal-cooked foods and industrial waste by products1. B (a) P is not manufactured and has no industrial use, but produced by industries involved in the production of aluminum, graphite, coal and asphalt and ubiquitously distributed throughout the environment. Human environmental exposure to B (a) P mainly occurs through cigarettes, ingestion of contaminated food and water1. Their levels in mainstream tobacco smoke are reported to be 20-40ng/cigarette 2, coke oven workers are exposed to about 42μg/m3 B (a) P3. Indoor exposure to B (a) P from cooking oil fumes has been reported to be 20μg/m3.4 Lioy et al., estimated that B (a) P intake ranged from 20-800ng/m3in people living in the vicinity of hazardous waste sites contaminated by PAHs 5. Similarly, B (a) P emitted from wood combustion in rural houses has been measured as high as 100μg/m3.6
Benzo (a) pyrene is generally large, flat molecule built from a collection of fused benzene-like rings and have a relatively low solubility in water, since they are rich in carbon and are hydrophobic. It can pass easily through the cell membranes and travel quickly into the cells. Benzo (a) pyrene does not attack DNA directly, but forms an intermediate within cells, with a reactive epoxide ring, for e.g. the 9, 10-epoxide (BPDE) that damages cellular macromolecules like proteins, lipids and DNA7. The bay region diol epoxides of PAHs are widely accepted as the ultimate carcinogenic forms of PAHs through their covalent binding to DNA. If this were so, one would expect that they would be carcinogenic at lower concentrations than the parental hydrocarbon. The formation and accumulation of B (a) P diol epoxide (BPDE)-DNA adducts, as represented by the common BPDE, are considered a critical early event in the initiation of carcinogenesis 8. In addition, the radical cationic forms of B (a) P may be involved in both the metabolism and metabolic activation leading to the formation of DNA adducts. However, the mechanism of the promotion stage remains unclear. Among the known biological molecules, lipids are considered to be extremely susceptible to the presence of reactive oxygen species (ROS). In particular, unsaturated fatty acids located in the neuronal membranes are prone to ROS attack, producing lipid peroxides 9. B (a) P is reported to disturb the antioxidant defense system and responsible to induce oxidative stress and has great potential for causing adverse effects due to redox cycling with their semiquinone radicals generating ROS, increasing oxidative stress and DNA damage. Antioxidants are critical in combating oxidative stress in neurons by scavenging free radicals. Induction of oxidative stress has been proposed previously as a possible mechanism of action for many pathological changes10.
B (a) P as an environmental neurotoxic compound
As the normal functioning of the brain is essentially dependent on an adequate oxygen supply to maintain energy metabolism, it is also vulnerable to oxidative damage, which can cause alterations in gene expression, impaired cellular signaling, and disruption of membrane integrity, altered neurotransmission and causing neuronal cell death11. Due to its high metabolic rate and relatively reduced capacity for cellular regeneration compared with other organs, the brain is believed to be particularly susceptible to the damaging affects of reactive oxygen species (ROS), which are derived from the metabolism of molecular oxygen12,13,14. The ROS decrease the antioxidant capacity or inhibit the antioxidant enzyme activity culminating in toxicant induced oxidative stress 15. These include its comparatively high oxygen utilization and hence generation of free radical by-products, its modest antioxidant defences, its lipid-rich constitution that provides ready substrates for oxidation, the reducing potential of certain neurotransmitters, and the presence of red ox-catalytic metals such as iron and copper 16. Additionally, the brain is also susceptible to secondary and self-perpetuating damage from oxidative cellular injury and the activated inflammatory response 17. In central nervous system (CNS), neurons derive their energy almost completely from oxidative phosphorylation in the respiratory chain of the mitochondria and adenosine triphosphate is generated by the reduction of oxygen to water through the sequential addition of four electrons and four protons. During this process, a leakage of high-energy electrons can potentially cause the formation of superoxide radicals 02- and through the action of superoxide dismutase (SOD), ultimately produce hydrogen peroxide (H202)18. Under conditions in which mitochondrial superoxide generation increases, or when antioxidant systems are depleted, H202 may accumulate and react with mitochondrial Fe2+, resulting in formation of reactive hydroxy! radicals (OH & OH») via Fenton reaction 19. Although H202 is not considered to be a free radical, it can easily form superoxide and hydroxyl radicals.
Normally, in healthy cells, there is a fine-tuned equilibrium between the generation of the reactive oxygen species and different enzymatic and non-enzymatic antioxidant defense systems. ROS normally exists in all aerobic cells in balance with biochemical antioxidants but, these have been shown to be modulated in diseases caused by free radical attack20. Oxidative stress occurs when this critical balance is disrupted by excess ROS production or a deficiency in an antioxidant system through B (a) P metabolism 21. Although the reason for this oxidative stress is not completely understood, it may be caused by the accumulation of toxic metabolites produced by B (a) P metabolism that leads to the excessive production of free radicals. Again an unusual increase in metabolic by-products directly, or indirectly, depletes a cell's antioxidant capacity. When a cell's pro-oxidants exceed its antioxidant capacity, free radicals accumulate and oxidative stress occurs (Fig.1). In brief, these free radicals play integral roles in cellular signaling, physiological immunological responses and mitosis. The resultant cellular damage may range from cellular structural damage and mitotic arrest, to apoptosis and cell necrosis, depending on the level of oxidative stress severity22_23.
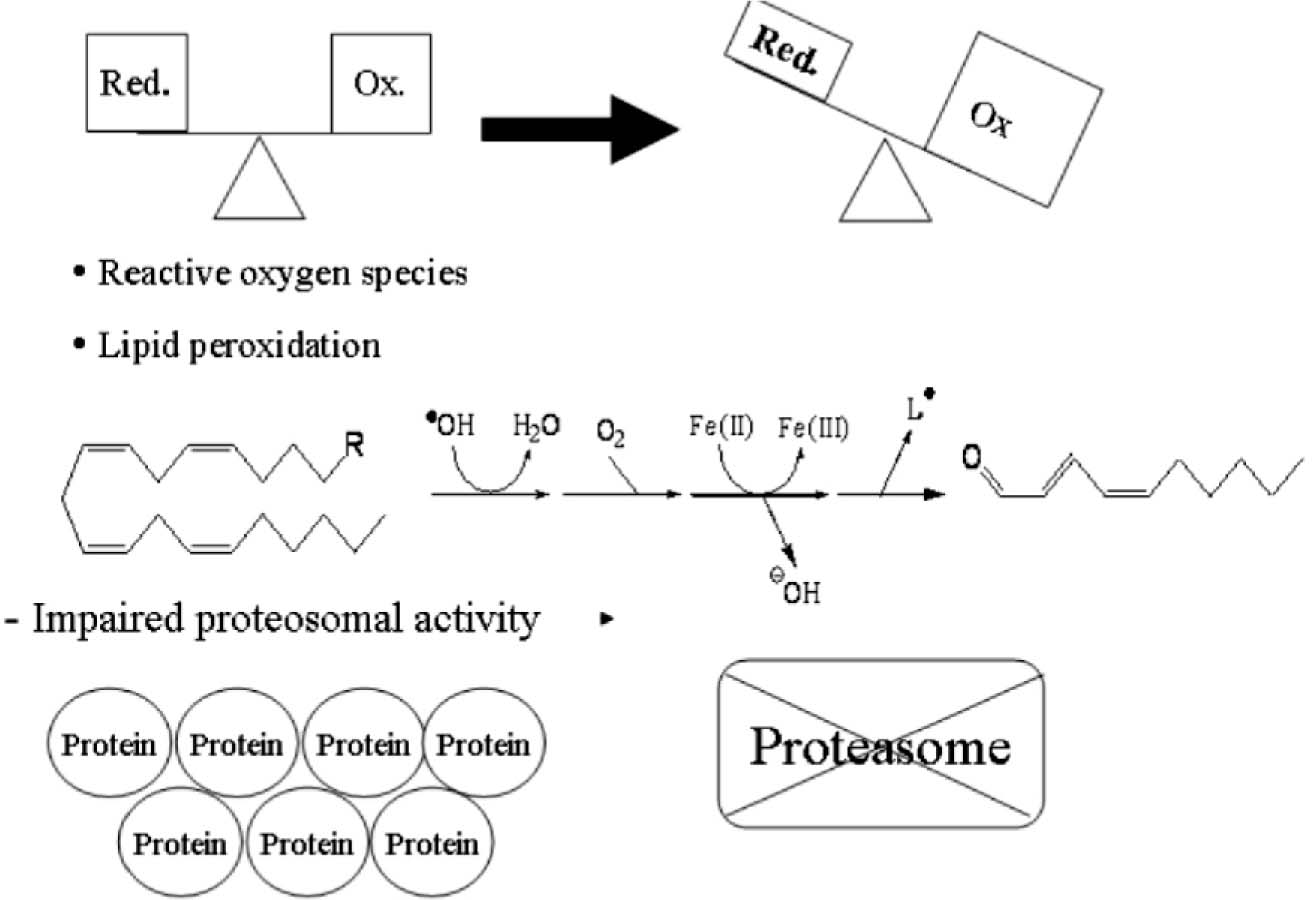
Figure 1. Oxidative stress vs. antioxidant defenses
Reactive oxygen Species ROS normally exists in all aerobic cells in balance with biochemical antioxidants. During B a P metabolism, when the bioactivation exceeds the detoxification it causes in production of biologically reactive metabolites, resulting in the formation of ROS. The increase in lipid peroxidation and decrease in antioxidant defense systems may lead to oxidative stress, sometimes result in insoluble protein an aggregate which fails to be degraded by the existing cellular machinery and accumulates with in the cytoplasm.
Induction of Cytochrome P450s (CYP) enzymes by B(a)P
The cytochrome P450 (CYP) are a superfamily of ubiquitous enzymes involved in the metabolism of a wide range of either endogenous or exogenous (xenobiotic) compounds. The elimination of PAHs, from organisms is mediated by enzymatic oxidation which the monooxygenase enzyme system with the CYPs as a functional link. CYP1A1 is an isoform that is highly induced by planar aromatic compounds like benzo (a) pyrene and which is able to metabolize a wide range of substrates, in particular PAHs. Benzo (a) pyrene, a model of PAH, undergoes a metabolic activation to form reactive intermediates before it is capable of inducing its mutagenic and carcinogenic effects in biological systems. The first step during B (a) P metabolism is to attach some hand-holds onto these slippery molecules like cytochrome P450. These cytochrome P450 enzymes add oxygen atoms to the rings, making them more water soluble and creating anchors for attachment of larger groups, like sugars or glutathione leading to their elimination. Unfortunately, some of the intermediate forms are highly dangerous and cause damage before they can be removed 24. However, there is convincing evidence suggesting that high CYP 1A1 activity could lead to toxicity e.g. during B (a) P metabolism. B (a) P when metabolized via Cytochrome P450s sometimes changes into B (a) P-7,8-oxide, which through hydration by epoxide hydrolase, is metabolized to B (a) P-trans-7,8-dihydrodiol [B (a) P-7,8-DHD]. B (a) P-7, 8-DHD may then serves as a substrate for a second CYP-dependent oxidation reaction, generating the ultimate carcinogenic metabolite B (a) P-7, 8-dihydroxy-9, 10-epoxide (BPDE), which could have profound effects on neurological functioning and other health related issues. In the nucleus, the BPDE may covalently bind to DNA, mainly forming deoxyguanoside-DNA adducts 7, which may result in misreplication and mutagenesis 25. The ratio between cytochrome P450 enzymes like CYP1A1 and phase II enzyme activities is critical to avoid the accumulation of putatively toxic reactive intermediates of B (a) P metabolism. B (a) P also act as both a substrate and inducer of this cytochrome P450 enzyme activity 26, which converts these substances into more polar, oxygenated products, facilitating their elimination from the cell. Another mechanism is the production of ROS, through which CYP1A1 could lead to toxicity by using B (a) P as a substrate 27. Hence, the physiological significance of the small amounts of this cytochrome present in brain microsome would depend on several properties like substrate specificity, inducibility, and distribution within the CNS. Thus, high CYP1A1 activity within the cell may be deleterious because of the generation of an intracellular oxidative stress and the subsequent oxidation of biological molecules. When the production of ROS is overwhelming, it will cause necrosis because of the irreversible degradation of cellular macromolecules and can induce apoptosis 28’29. However, some of the intermediates generated during B (a) P metabolism are chemically reactive, electrophilic derivatives which can be more toxic than the parent compound 30.
In addition to effects of the PAH metabolites, there is another aspect of the action of this class of carcinogens caused by interaction of a nonmetabolised compound with the cytoplasmic Ah-receptor (Aromatic hydrocarbon-receptor). Although, B (a) P itself is not genotoxic, its biological effects are initiated by binding to a ligand-dependent transcription factor termed aromatic hydrocarbon receptor (AhR) 31. This ligand-bound AhR is translocated to the nucleus and forms a heterodimer with aryl hydrocarbon receptor nuclear translocator (ARNT). After metabolic activation within the cells, some active metabolites of B (a) P trigger to form Ligand-activated AhR-ARNT complexes and can interact with specific promoter elements xenobiotic-response elements (XREs) to initiate the transcriptional activation of many genes, including members of the cytochrome P450 enzymes such as CYP1A1, CYP1A2 and CYP1B1 32. Mainly, Phase I enzymes are responsible for the production of aryl hydrocarbon hydrolase (AHH), and the oxidative metabolism of AhR ligands. B (a) P is metabolized by AHH to a procarcinogen compound BPDE, (+-)-anti-7β, 8α-dihydroxy-9α, 10a-epoxy-7, 8, 9, 10-tetrahydropyre), which binds to DNA and forms predominantly covalent (+) trans adducts at the N2 (N2) position of guanine 33. Phase I enzymes also increase the production of reactive oxygen species (ROS)34, which have been shown to be associated with lipid peroxidation, oxidative DNA damage and other pathological effects 35. The biochemical and molecular studies by Nebert et al.,31also established that AhR plays a key role in cell-cycle regulation and apoptosis. AhR activation by PAHs including B (a) P leads to the induction of AHH, which generate reactive metabolites from the parent compound (by the AhR regulation of AHH enzyme), and which contribute to apoptosis and other cellular damage in biological system.
Studies performed in in vitro have demonstrated clearly that CYP1A1 is involved in the metabolic activation of B (a) P into reactive intermediates, rather than the non-metabolised parent compound, and is responsible for B (a) P-mediated mutations, cancer and birth defects 36. Among the various forms of P450 determined so far, CYP1A1 and CYP1B1 have been shown to be the most important human P450 enzymes in the metabolic activation of PAHs and PAH dihydrodiols 37. Several cytochrome P450 enzymes are associated with key steps in the oxidation of B (a) P, namely 7, 8-epoxidation of B (a) P and 9; 10-epoxidation of B (a) P-7, 8-diol and CYP1A1 has been demonstrated to be the most active in these oxidations in mammals38. Uncoupling of electron transfer and oxygen reduction from monoxygenation by CYP1A1 and CYP1A2 can result in the release of O2-, H2O2 and OH’39. These reactive oxygen species or oxyradicals react with DNA, proteins and membrane lipids in the intracellular milieu31,40 thereby contribute to cytotoxic and neurological deficits. Many of the recent reports primarily focus on induction of CYPs40,42; however, the mechanism by which the B (a) P acts on CYP450 metabolic activity has not been investigated.
Neurological effects after exposure to B(a)P
There is growing evidence that prenatal exposure to air pollutants from combustion of coal and other fossil fuels have adverse effects on fetal growth and early child neurodevelopment (Fig.2). Recent studies by Saunders et al, also demonstrated that oxidative stress in the CNS through the generation of reactive oxygen species and repression of enzymatic antioxidants may be a critical mechanism in the behavioral effects induced by B (a) P21. Furthermore, it has been reported that there is close relationship between oxidative stress and locomotor behavior and striatal function, and also between hippocampal oxidative stress and age-induced cognitive decline 43,44. Molecular and epidemiological research has shown that fetuses and infants are more susceptible than adults to the harmful effects of a variety of environmental contaminants, including PAHs 45. Experimental animal studies have demonstrated that B (a) P, is a toxicant and produces a variety of neurodevelopmental effects as a result of nervous system damage, including decreased motor activity; neuromuscular, physiologic, and autonomic deficits and decreased responsiveness to sensory stimuli 46-48. It was also reported that gaiting, loss of coordination, neuromuscular weakness, decrease response to sensory motor stimuli, increased urination and defecation were demonstrated following acute exposure to B (a) P and fluranthene, a closely related PAH compound 21. They showed inhibition of motor activity in rats exposed to 27μg/m3of B (a) P by inhalation21. According to Perera ei a/., DNA-adducts_were associated with reduced DQs in the motor and language areas and also associated with increased odds of developmental delay in the motor area 49.
In epidemiologic studies, prenatal exposure to PAHs has been shown to be associated with reduced birth weight and head circumference 50–53. In the present cohort, reduction of head circumference was associated with PAH-DNA adducts in cord blood52. Reduction of weight or head circumference at birth has been correlated with lower IQ as well as poorer cognitive functioning and school performance in childhood 51. In some studies prenatal exposure to inhaled B (a) P was reported to cause deficits in 'learning and memory' as revealed by the fixed-ratio performance of behavioral study and long term potentiation47,54. In Wistar rats cognitive deficits have been reported after administration 25mg/kg i.p. dose of 3-methylcholanthrane, another PAH compound53,54. According to Cardozo et al unequaled oxidative stress in brain regions has some impact on spontaneous motor activity and cognition11. Several studies have associated PAH exposure with decrements in the mental development index on the Bayley Scales of Development at 3 years of age 48. The deficits in development at 2-3 years of age may be educationally meaningful because compromised function at an early age may have a negative impact on subsequent school performance 55. Others have also suggested oxidative changes, such as cumulative oxidative DNA damage, to be a common pathophysiological mechanism underlying major depression and medical co-morbidities 56.
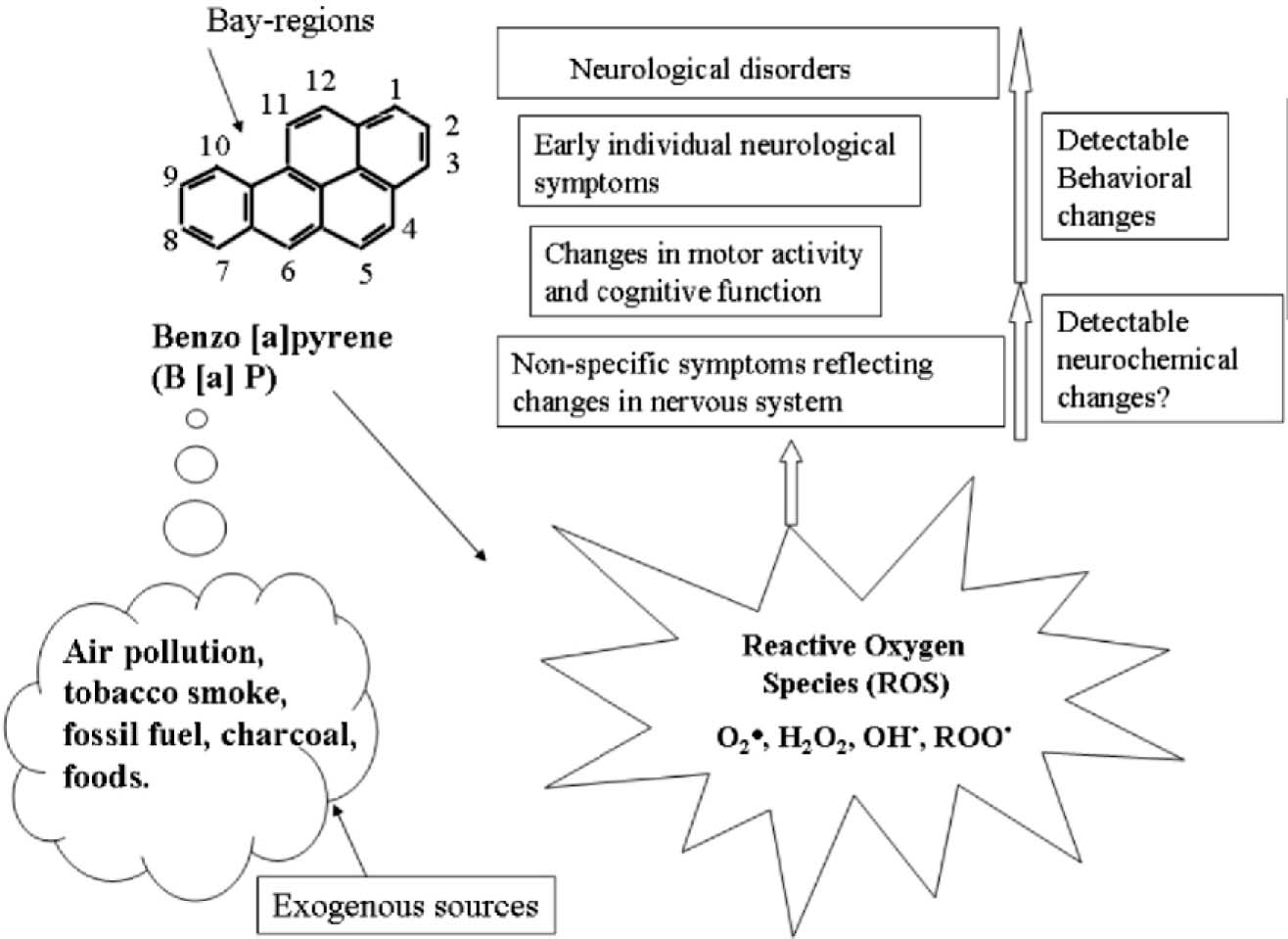
Figure 2. Potential behavioral neurotoxicity of benzo a pyrene
Human exposure to B a P essentially takes place through the ingestion or by inhalation of particulates in the air, cigarette smoking etc. The CNS may be extremely susceptible to attack by ROS reactive oxygen species derived from B (a) P metabolism, which may results repression of enzymatic antioxidants causing in behavioral changes and altering the biochemical's in brain.
Benzo (a) pyrene metabolism and oxidative stress
Oxidative stress is believed to be one of the major causes of many human diseases as it can result in severe cellular dysfunction due to peroxidation of membrane lipids, protein modification, depletion of nicotinamide nucleotides, cytoskeletal disruption and DNA damage 57. It has been implicated as an important mechanism in the carcinogenicity of PAHs 10. Oxidative and genotoxic stress induced by PAHs, including B (a) P, activate check point mechanisms for cell cycle control and apoptosis in mammalian cells 31. It was reported that most of these chemicals induce free radical-mediated lipid peroxidation leading to disruption of biomembranes and dysfunction of cells and tissues 49. It is a deleterious process that can be an important mediator of damage to cell structure and consequently have an important role in the etiology of various disease states. Some environmental chemicals, like B (a) P when led into oxidative stress, it mainly affect the rates of metabolism, growth and development, higher nervous function, as well as ability to deal with stress due to the antioxidant defence mechanism of a cell or tissue, resulting in some abnormalities. Our brain always operates using a highly intricate chemical communication system and for this to occur properly a receptor must have an affinity for specific chemical ligands or signals, to initiate a response. As brain encounters high levels of oxidative stress as it consumes ~20 % of the inhaled oxygen and possesses low levels of antioxidant enzymes then the metabolism of B (a) P via Cytochrome P-450 generates free radicals, which can disrupt the intracellular oxidant/ antioxidant balance 58. However, the question whether uncontrolled formation of ROS species is a primary cause of down stream consequence of the pathological process. Because neurodegenerative conditions like Alzheimer's disease, Parkinson's diseases and Huntington's disease have oxidative stress implication in their pathogenesis 28. In several recent reviews, the role of oxidative stress and oxidative damage to biomolecules has been supported by the pathogenesis of neurodegenerative disease, and specifically e.g. Alzheimer's disease 59.
Generally, three pathways have been proposed by researchers to explain B (a) P metabolism (Fig.3.). The first pathway involves the formation of epoxides, catalyzed by CYP-dependent m on oxygenases. Further metabolism includes hydration by microsomal epoxide hydrolase to diols that are oxidized by CYP to produce a diol epoxide, for e.g. the B (a) P-7,8-dihydrodiol, 10-epoxide (BPDE). The BPDE is the main metabolite that causes toxicity and carcinogenicity by covalently binding to the guanine residue on DNA to form DNA-adducts. The second pathway involves a one electron oxidation of B a P to 6-oxo-B (a) P-radical intermediates that may attack DNA resulting in depurination 8. The third pathway involves enzymatic dehydrogenation of dihydrodiol metabolites to yield quinone intermediates that may combine directly with DNA or generate reactive oxygen species, capable of attacking DNA 60. Therefore, quinone formation is associated with events in the B (a) P metabolism. Of the three pathways, the epoxide pathway is the predominant pathway of B (a) P metabolism (Fig.3), and the vulnerability of neural tissues to B (a) P-induced toxicity depends on the CYT P-450-dependent metabolic capacity of the tissues 36. Further, dihydrodiol dehydrogenase and peroxidase were reported to be involved in the metabolic conversion of B (a) P to reactive and redox active o-quinones, that have been demonstrated in chemical systems to undergo one electron redox cycling with their semiquinone radicals resulting in the formation of ROS and lipid peroxidation 61,62. The increase in lipid peroxidation and subsequent decrease in antioxidant defence systems may contribute to an increased susceptibility to oxidative stress 63.The increased formation of the dihydrodiols also indicate the tilt of B (a) P metabolism towards toxification and the decreased formation of this metabolite group, points shifting the balance towards detoxification 21. Therefore, bioactivation of B (a) P to highly reactive metabolites and decreased levels of antioxidants enzymes resulting in oxidative stress might be the cause for η euro behavioral toxicity. Continued research is needed to better understand the mechanisms and specific pathways involved in ROS-induced cell death, and to determine the most rational and effective combination of redox-active agents, resulting in some neurobehavioral changes.
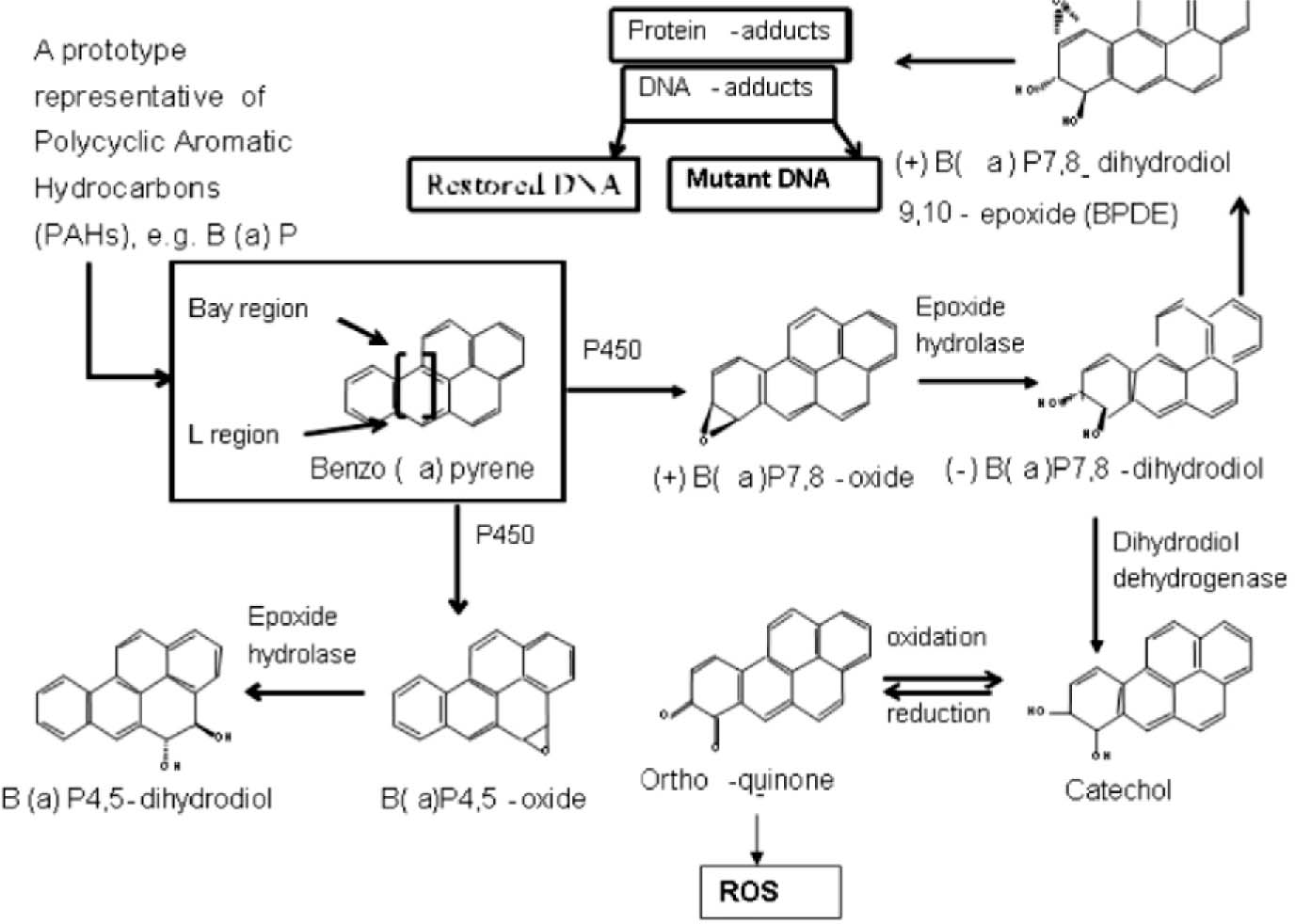
Figure 3. Metabolic pathways of Benzo a pyrene
In the profile of B a P metabolism, major primary and secondary metabolites of B a P a re formed as a result of oxidation of the parent compound by the cytochrome P450 enzymes. The epoxides and hydroxyl metabolites, with further oxidation results the formation of quinines, diols, and diol epoxides, are mutagenic. These three predominant quinines can affect the redox cycle between their hydroquinone and semiquinone intermediates to generate reactive oxygen species ROS such as superoxide anion, H202 and hydroxyl radicals by Fenton chemistry, which can lead to cytotoxicity.
Oxidative stress and neurological disorders
The theory of oxidative stress as a pathophysiological mechanism, at its most basic, can be explained by the concept. Oxygen paradox that while oxygen is essential for aerobic life, excessive amounts of its free radical metabolic by-products are toxic 22. The brain does not have high levels of protective enzymes and the inability of adult neuronal cells to replicate and replace damaged DNA, and owing to lower level of glutathione peroxidase (GSH-Px), catalase (CAT) and having high levels of SOD 25,64. Glutathione exists in the reduced (GSH) and the oxidized (GSSG) forms, which can be inter-converted by the enzymes glutathione peroxidase (Gpx) and glutathione reductase (GR). In mammalian cells, the cycling between GSH and GSSG serves to remove reactive oxygen species (ROS), to protect the cells from oxidative stress 65. A proper balance among these enzymes is required for an effective antioxidant defense because, excess SOD in relation to GSH-Px and CAT, enzymes of peroxide metabolism, contribute to brain pathology 66. Depletion of GSH is associated with number of human diseases including Parkinson's disease, Alzheimer's disease 67. The increased SOD activity was associated with manic and depressive episodes, whereas another study found a trend for reduced SOD in bipolar disorder and significantly reduced CAT levels from studies with patient samples that include other psychiatric disorders 68,69. An increase in the lipid peroxidation product, TBARS, was also reported for both bipolar disorder and schizophrenia70. Some of the studies suggested that 02” causes changes in intracellular calcium by affecting microsomal and mitochondrial calcium stores and thereby leading to changes in signal transduction and differential gene expression. One possibility is that damage to these organelles can contribute to decreased energy production and increased levels of intracellular free calcium which, in turn can lead to cellular dysfunction and even death. Dysregulation of secondary messenger calcium has been described in bipolar disorder in response to oxidative stress71.
Sometimes, brain is vulnerable to oxidative damage due to its high oxygen utilization, its high content of oxidisable polysaturated fatty acids (PUFAs), and the presence of redox-active metals copper and iron. The redox state of the cell is largely linked to these redox couple and is maintained within strict physiological limits. Mainly, iron regulation ensures that there is no free intracellular iron; however in vivo, under stress conditions an excess of superoxide releases “free iron” from iron containing molecules. The entry and release of iron from iron-storage protein, ferritin, occurs via the “free iron ferrous labile pool”, active in Fenton chemistry.
Besides superoxide, 6-hydroxydopamine a neurotoxin implicated in Parkinson's diseases can release ferritin iron also. There is also strong experimental evidence showing that oxidative stress and lipid peroxidative products can cause decreases in dopamine and inhibit Na+/K+ ATPase activity as well72. In humans, only a handful of relevant studies have been published. These reported elevated lipid peroxidation products and antioxidant changes in obsessive-compulsive panic disorder and social phobia70,73,74. A study of anxious women found reduced total antioxidants capacity when compared with non-anxious control group along with other parameter like impaired immune functioning 75. The notion of oxidative stress mechanism underlying anxiety disorder has been known for some years which might play a major role in setting up a vicious etiological cycle involving free radicals, inflammatory cytokines in post traumatic stress disorder 29. However oxidation biology research in anxiety disorder is still at its infancy and the bulk of limited literature is insufficient to generate intriguing findings.
Recent studies have also indicated that ROS play a key role in the pathophysiological pathway of wide variety of clinical and experimental diseases 26. About 100 different types of disorders, like rheumatoid arthritis, hemorrhagic shock, cardiovascular diseases, gastro-intestinal ulcerogenesis and AIDS, have been reported as the ROS mediated disorders16,17,76. Some brain related specific examples of ROS-mediated diseases are Alzheimer's disease and Parkinson's disease 15,76. In neurodegenerative diseases like Parkinson's, Alzheimer's and amyotrophic lateral sclerosis ALS, ROS damage has been reported within the specific brain region that undergo selective neurodeganeration. Protein oxidation has been reported in the hippocampus and neocortex of patients with Alzheimer's diseases, Lewy bodies in Parkinson's disease and within the motor neurons in ALS 77. Lipid peroxidation has also been identified in the cortex and hippocampus of patients with Alzheimer's disease, substantia nigra of patients with Parkinson's disease and spinal fluid in patients with ALS. It is also known that ROS can cause neuron and astrocyte death through apoptosis and necrosis, but, B (a) P-induced apoptosis have not been fully investigated. Innate deregulation of apoptosis and oxidative processes has been suggested by a recent study, in which the hippocampal expressions of genes encoding DNA repair and antioxidant enzymes were found to be down-regulated in bipolar disorder, while many apoptosis genes were up-regulated 78. According to Chung et al., B (a) P metabolites produced by Cytochrome P450 enzymes can elicits genetic toxicity by forming DNA adducts and results in DNA damage-induced apoptosis 79. Disorders in DNA repair typically cause additional symptoms such as mental retardation, photosensitivity, immunodeficiency and neoplasia 80.
Oxidative stress is also related to glutamate release and NMDA receptor activation during cerebral ischemia-reperfusion, production of 02~ in neurons, brain macrophages and glutamate-induced ROS production in astrocytes. Recently, Grova et al., demonstrated that chronic exposure to B (a) P in adult mice modulates gene expression of NMDA NR1 subunit in brain areas that are highly involved in cognitive function like the hippocampus 81-83. Evidences implicating ROS in major degenerative disease is also consistent with their role in brain aging 84. Thus exposure to various environmental chemicals like B (a) P acting through oxidative stress and the reduced capacity for the homeostatic maintenance of synaptic plasticity mechanisms during brain development may contribute to subsequent behavioral learning deficits. The exact mechanism by which B (a) P administration causes reduction in antioxidant enzymes is not clear 21,85. Thus, B (a) P-mediated toxicity might be due to its own oxidative properties, its reactive metabolites or both. It is not known whether B (a) P-mediated toxicity is due to its direct toxicity, the toxicity of metabolites or both.
Conclusions and future prospects
Many questions remain unanswered with regard to the role of B (a) P in nervous system dysfunction. Is B (a) P a full carcinogen? What is the major role of ROS and what form of the compound (original or metabolized) is acting? Whether there are changes in functioning of the same regulatory systems as in by nongenotoxic mechanism? Taken together, the study reviewed herein supports the view that an imbalance between oxidants and antioxidants and between proteases and antiproteases may play an important role in the susceptibility of CNS. It becomes clear that reduced antioxidant potential which might result from the binding of free radicals to the active sites of these enzymes, contribute to the increased oxidative stress that is associated with nervous system dysfunction. This ROS production could contribute to repress the expression of oxidative-stress-sensitive genes. The negative regulation of transcription by ROS could affect several CYP isoforms, exhibiting high CYP metabolism, which would rapidly activate the pro-apoptotic machinery leading to a massive cell death following different pathways in order to determine the cell fate. Another potential mechanism for the alterations in enzyme status might be through transcription factors causing changes in antioxidant gene expression. B (a) P is a carcinogen, which induces both the initiation and promotion stages of carcinogenesis. The mechanism of the initiation is well studied while the promotion stage of the carcinogenic action is virtually uninvestigated. We await more experiments on oxidative stress in the CNS through generation of ROSs and the change in enzymatic antioxidants, which may be the critical mechanism in neurological diseases, induced by B (a) P. These chemicals are affecting the normal growth and development at a degree far greater than ever imagined and it needs some of the challenging areas for further research in oxidative stress related diseases in brain. It will be necessary to examine the relationships between metabolite formation of PAHs and oxy-PAHs and DNA adduct formation and metabolic enzyme induction by the processes in detail. Further, more laboratory studies aimed at identifying the underlying mechanisms of B (a) P exposure, particularly on those individuals deemed to be a greatest risk, and are badly needed.
Acknowledgements
Financial assistance of DBT, ICMR, DST and
CSIR is gratefully acknowledged.
Competing interests - None,
Source of Funding-DBT, DST, ICMR, CSIR
Received Date : 19 March 2009
Revised Date : 1 April 2009 Accepted Date : 20 April 2009
References
1 Agency for Toxic Substances and Disease Registry (ATSDR). Toxicological Profile for Polycyclic Aromatic Hydrocarbons (PAHs). US Department of Health & Human Service: Atlanta, GA. 1995.
2 Kuller LH, Garfield L, Correa P, et al. Contribution of passive smoking to respiratory cancer. Environ Health Perspect 1986; 70:57-69.
3 Lewtas J, Walsh D, Williams R, et al. Air pollution exposure-DNA adduct dosimetry in humans and rodents: evidence for nonlinearity at high doses. MutatRes1997;378:51-63.
4 Chiang TA, Wu PF, Wang LF, et al. Mutagenicity and polycyclic aromatic hydrocarbon content of fumes from heated cooking oils produced in Taiwan. Mutant Res 1997; 381:157-61.
5 Lioy PL, Waldman JM, Greenberg A, et al. The total Human Environmental Exposure Study (THEES) to benzo(a)pyrene: comparision of the inhalation and food pathways. Arch Environ Health 1988; 43:304-12.
6 Viau C, Hakizimana G, Bouchard M. Indoor exposure to PAHs and carbon monoxide in traditional houses in Burundi. Int Arch Occup Environ Health 2000;73:331-338.
7 Dipple A, Moschel RC, Bigger CAH. Polynuclear aromatic hydrocarbons. In: Searle CE, Ed., Chemical Carcinogens, vol 1. American Chemical Society Washington, DC 1984; 41-163.
8 Cavalieri KK, Rogan EG. Central role of radical cations in metabolic activation of polycyclic aromatic hydrocarbons. Xenibiotica 1995; 25:677-88.
9 Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement and significance. Am J Clin Nutr 1993; 57: 715-25.
10 Andersson HE, Lindqvist R, Westerholm K, et al. Neurotoxic effects of fractionated diesel exhausts following microinjection in rat hippocampus and striatum. Environ Res 1998; 76:41 -51.
11 Cardozo PF, Song S, Parthasarthy A, et al. Oxidative DNA damage in the aging mouse brain Mov Disord 1999; 14:972-80.
12 Halliwell B, Gutteridge JM. Role of free radicals and catalytic metal ions in human disease: an overview Methods Enzymol 1990; 186:1-85.
13 Halliwell B. Oxygen and nitrogen are procarcinogens. Damage to DNA by reactive oxygen, chlorine and nitrogen species: measurement, mechanism and the effects of nutrition. Mutat Res 1991;443:37-52.
14 Halliwell B, Chirico S. Lipid peroxidation: its mechanism, measurement and significance. Am J Clin Nutr 1993; 57: 715-25.
15 Patel S, Sinha A, Parmar D. An update on the role of environmental factors in Parkinson's disease. Annals of Neurosciences 2005;12(4):79-86.
16 Halliwell B, Gutteridge JMC. Free radicals in biology and medicine. Oxford Clarendon Press, 1999.
17 Halliwell B. Oxidative stress and neurodegeneration: where are we now? Journal of Neurochemistry 2006; 97:16621-26.
18 Fridovich I. Superoxide anion radical (02),superoxide dismutases and related matters. J Biol Chem 1997; 272; 18: 515-17.
19 Sutton HC, Winter bourn CC. On the participation of higher oxidative states of iron and copper in Fenton reactions. Free Radic Biol Med. 1989; 6: 53-60.
20 NikiE. Antioxidant defense in eukaryotic cells: an overview, In: G Poli E Albano MU Dianzani (Eds.), Free Radicals: From basic science to Medicine, Birkahauser, Basel, Switzerland, 1993; 365-373.
21 Saunders CR, Das SK, Ramesh A, et al. Benzo(a) pyrene-induced acute neurotoxicity in the F-344 rat: role of oxidative stress. J Appl Toxicol 2006; 26:427-38.
22 Davies KJ. Oxidative stress, antioxidant defences, and damage removal, repair and replacement systems. IUBMB Life 2000; 50:279-89.
23 Finkel T, Holbrook NJ. Oxidants, oxidative stress and the biology of ageing. Nature, 2000; 408:239-47.
24 Evans MD, Griffiths HR, Lunce J. Reactive oxygen species and their cytotoxic mechanisms of cell toxicity In: Chipman JK (ed) JAI Press Inc. London,1997; 25-73.
25 Anand A, Prabhakar S, Kaul D. Genetic polymorphism in muscle biopsies of Duchenne and Becker muscular dystrophy patients. Neurol India 1999;47(3):218-23.
26 Cohn JA, Alvares AP, Kappas A. On the occurrence of Cytochrome P-450 and aryl hydrocarbon hydroxylase activity in rat brain. Journal of Experimental Medicine 1977; 5:1607-11.
27 Puntarulo S, Cederbaum Al. Production of reactive oxygen species by microsomes enriched in specific human cytochrome P450 enzymes. Free Radic Biol Med 1998;24:1324-30.
28 Bolotina NA, Gasparian AV, Dubovaja TK, et al. Benzo(a) pyrene-Dependent Activation of Transciption Factors NF-kB and AP-1 Related to Tumor Promotion in Hepatoma Cell Cultures. Biochemistry (Moscow).72:682-89.
29 Shukla R. Nitric oxide in neurodegeneration.Annals of Neurosciences 2007;14 (1):13-20.
30 Miller CS. Are we on threshold of a new theory of disease? Toxicant-induced loss of tolerance and its relationship to addiction and abdiction. Toxicology and Industrial health, 1999; 15:284-94.
31 Hankinson O. The aryl hydrocarbon receptor complex. Annu Rev Pharmacol Toxicol 1995;35:307-40.
32 Nebert DW, Roe AL, Dieter MZ, et al. Role of the aromatic hydrocarbon receptor and Ah gene battery in the oxidative stress response, cell cycle control and apoptosis. Biochem Pharmacol 2000; 59:65-85.
33 Bahadur G, Ling KL, Katz M. Statistical modeling reveals demography and time are the main contributing factors In global sperm count changes between 1938 and 1996. Human Reproduction; 1996; 11:2635-39.
34 Lopes S, Jurisicova A, Sun JG, Casper RF. Reactive oxygen species: potential cause for DNA fragmentation in human spermatozoa. Hum Reprod 1998; 13: 896-900.
35 Park JY, Shigenaga MK, Ames BN. Induction of cytochrome P4501A1 by 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin or lndolo(3,2-b)carbazole is associated with oxidative DNA damage. Proc Natl Acad Sci U S A, 1996; 93:2322-7.
36 Miller KP, KS Ramos. Impact of cellular metabolism on the biological effects of benzo(a)pyrene and related hydrocarbons. Drug MetabRev. 2001; 33:1-35.
37 Shimada T, Oda Y, Gillam EMJ, et al. Metabolic activation of polycyclic aromatic hydrocarbons and other procarcinogens by cytochrome P4501A1 and P450 1B1 allelic variants and other human cytochrome P450 in Salmonella typhimurum. Drug Metab Dispos 2001; 29:1176-82.
38 Scandalios JG. The rise of ROS. Trends Biochem.Sci. 2002; 27:483-486.
39 Dugan LL, Choi DW. Excitotoxicity, free radicals and cell membrane changes, Ann Neurol 1994;35:17-21.
40 Zangar RC, Davydov DR, Verma S.Mechanisms that regulate production of reactive oxygen species by cytochrome P450. Toxicol Appl Pharmacol 2004; 199:316-31.
41 Mahadevan B, Marson CP, Dashwood WM, et al. Effects of a standard complex mixture derived from coal tar on the metabolic activation of carcinogenic polycyclic aromatic hydrocarbon in human cells in culture. Chem Res Toxicol 2005; 18:224-31.
42 Marston CP, Pereira C, Ferguson J, et al. Effects of a complex environmental mixture from coal tar containing PAHs on the tumor initiation, PAH-DNA binding and metabolic activation of carcinogenic PAH in mouse epidermis. Carcinogenesis. 2001; 22:1077-86.
43 Kumar P, Padi SSV, Naidu PS. Protective Effect of Antioxidants on 3-Nitropropionic Acid Induced Oxidative Stress and Cognitive Impairment. Annals of Neurosciences 2006; 13(2): 41 -47.
44 Nicolle MM, Gonzalez J, Sugaya K, et al. Singatures of hippocampal oxidative stress in aged spatial learning-impaired rodents. Neuroscience 2001; 107: 415-31.
45 Perera FP, Tang D, Whyatt RM., et al. DNA damage from polycyclic aromatic hydrocarbons measured by benzo(a)pyrene-DNA adducts in mothers and newborns from Northen Manhattan, the World Trade Center Area , Poland, and China. Cancer Epidemiol Biomarkers Prev 2005; 14: 709-14.
46 Archibong AE, Inyang F, Ramesh A, et al. Alteration of pregnancy related hormones and fetal survival in F-344 rats exposed by inhalation to benzo (a) pyrene. Reprod Toxicol 2002; 16: 801-08.
47 Saunders CR, Ramesh A, Shockley DC. Modulation of neurotoxic behavior in F-344 rats by temporal disposition of benzo a) pyrene. Toxicol Lett 2002; 1291:33-45.
48 Wormley D, Chirwa S, Zhang W, et al. Prenatal exposure to dioxin and inhaled benzo a pyrene: reduced capacity for long-term potentiation in the F1 generation. The Toxicologist 2003; 72:1:125.
49 Perera FP, Rauh V, Whyatt RM, et al. Effects of prenatal exposure to airborne polycyclic aromatic hydrocarbons on neurodevelopment in the first 3 years of life among inter-city children. Environ Health Perspect 2006; 114:1287-92.
50 Choi Jedrychowski, W Spengler J Camman DE, et al. International studies of prenatal exposure to polycyclic aromatic hydrocarbons and fetal growth Environ Health Perspect 2006; 114:1744-50.
51 Sram RJ, Binkova B, Dejmek J, et al. Ambient air pollution and pregnancy outcome: a review of the literature. Environ Health Perspect 2005; 113:375-82.
52 Tang D, Li TY, Li JJ, et al. PAH-DNA Adducts in cord blood and fetal and child development in a Chinese cohort. Environ Health Perspect 2006; 114: 1297-1300.
53 Perera FP, Whyatt RM, Jedrychowski W, et al. Relationship between polycyclic aromatic hydrocarbon DNA-adducts, environmental tobacco smoke, and child development in the World Trade Center cohort. Environ Health Perspect 2007;115:1497-1502.
54 Wu J, Ramesh A, Nayyar T, et al. Assessment of metabolites and Ahr and CYP1A1 mRNA expression subsequent to prenatal exposure to inhaled benzoapyrene. Int. J. Dev. Neurosci. 2003; 21:33346.
55 Konstandi M, Pappas P, Johnson E, et al. Suppression of the acquisition of conditioned avoidance behavior in the rat by 3-methyl-choanthrane. Pharmacol Biochem Behav. 1997; 56: 637-41
56 Drillen CM, Pickering RM, Drummond MB. Predictive values of screening for different areas of development. Dev Med Child Neurol 1988; 30:294-305.
57 Forlenza MJ, Miller GE. Increased serum levels of 8-hydroxy-2'-deoxyguanosine in clinical depression. Psychosomatic Medicine 2006; 68,1-7.
58 Scandalios JG. The rise of ROS. Trends Biochem. Sci. 2002; 27:483-486.
59 Barzilai A, Rotman G, Shiloh Y. ATM deficiency and oxidative stress: a new dimension of defective response to DNA damage. DNA repair 2002; 1,3-25.
60 Smith MA, Rottkamp CA, Nunomura A, et al. Oxidative stress in Alzheimer's disease Bio-chem Biophys Acta 2000; 1502: 139-144.
61 Penning TM, Burczynski ME, Hung CF, et al. Dihydrodiol dehydrogenases and polycyclic hydrocarbon activation: generation of reactive and redox active o-quinones. Chem. Res Toxicol 1999; 12:1-18.
62 Mulder PP, Devanesan P, Van Alem K, et al. Flurobenzoapyrene as probes of the mechanism of cytochrome P450-catalyzed oxygen transfer in aromatic oxygenations. Free Radic Biol Med 2003;34:734-45.
63 Casetta I, Govoni V, Granieri E. Oxidative stress, antioxidants and neurodegenerative diseases. Curr Pharm Des 2005; 11:2033-52.
64 Badary OA, AbdEI-Gawad H M,Taha RA, et al. Behavioral and neurochemical effects induced by subchronic exposure to 40 ppm toluene in rats. Pharmacol. Biochem Behav 2003; 74:997-1003.
65 Cohen G, Greenwald RA (Eds.). Oxyradicals and Their Scavenger Systems. Vol.1. Molecular Aspects. Proc. Third Intl. Conf. on Superoxide and Superoxide Dismutase. Elsevier Publishing Co. Inc. Ellenville,NewYork,1983.
66 Jones DP. Redox potential of GSH/GSSG couple: assay and biological significance. Methods Enzymol. 2002; 348:93-112.
67 Groner Y, Elroy-Stein O, Avraham, KB, et al.Down syndrome clinical symptoms are manifested in transfected cells and transgenic mice over expressing the Human Cu/Zn-superoxide dismutase gene. J Physiol 1990; 84:53-77.
68 Wu MT LH,Lee CK, Ho SC, et al. Environmental exposure to cooking oil fumes and cervical intraepithelial neoplasm. Environ Res. 2004; 94:25-32.
69 Andreazza AC, Cassini C, Rosa AR, et al. Serum S100B and antioxidant enzymes in bipolar patients. Journal of Psychiatric Research 2007; 41:523-29.
70 Ranjekar PK, Hinge A, Hedge MV, et al. Decreased antioxidant enzymes and membrane essential polyunsaturated fatty acids in schizophrenic and bipolar mood disorder patients. Psychiatry Research, 2003; 121:109-22.
71 Kuloglu M, Ustundag B, AtmacaM.et al Lipid peroxidation and antioxidant enzyme levels in patients with schizophrenia and bipolar disorder. Cell Biochemistry and Function, 2002; 20:171 -175.
72 Berk M, Kirchmann NH, Butkow N. Lithium blocks 45Ca2+ uptake into platelets in bipolar affective disorder and controls. Clinical Neuropathology 1996; 19: 48-51.
73 Madrigal J, Moro M, Lizasoain I, et al.Induction of cyclooxygenase-2 accounts for stress-induced oxidative status in rat brain. Neuropyscho-pharmacology 2003; 28:1579-88.
74 Ersan S, Bakir S, Erdal Ersan E, et al.Examination of free radical metabolism and antioxidant defence system elements in patients with obsessive-compulsive disorder. Progress in Neuropsychopharmacology and biological Psychiatry 2006; 30, 1039-42.
75 Atmaca M, Tezcan E, Kulugo M, et al. Antioxidant enzyme and malonaldehyde values in social phobia before and after citalopram treatment. European Archives of Psychiatry and Clinical Neuroscience 2004; 254: 231-35.
76 Shukla R. Nitric oxide in neurodegeneration. Annals of Neurosciences 2007;14(1): 13-20.
77 Siddiqui MA, Kashyap MP, Khanna VK, et al. Metabolism of 4-hydroxy trans 2-nonenal (hne) in cultured pc-12 cells. Annals of Neurosciences 2008;15(3): 60-68.
78 Andersen, JK. Oxidative stress in neurodegeneration: cause or consequences? Nat. Rev Neurosci. 2004;18-25.
79 Benes FM, Matzilevich D.Burke RE, etal. The expression of proapoptosis genes is increased in bipolar disorder, but not in schizophrenia. Molecular Psychiatry 2006;11:241-251.
80 Chung JY, Kim JY, Kim YR, etal. Abundance of aryl hydrocarbon receptor potentiates benzo (a) pyrene-induced apoptosis in Hepa1c1c7 cells via CYP1A1 activation. Toxicology 2007; 235:62-72.
81 De Boer J. ND, Hoeijimakers JH. Nucleotide excision repair and human syndrome. Carcinogenesis, 2000; 21:453-460.
82 Grova N, Valley A, Turner ID,etal. Modulation of behavior and NMDA-RI gene mRNA expression in adult female mice after sub-acute administration of Benzo (a) pyrene. Neurotoxicology; 2007;28: 630-36.
83 Haining Rl, Nichols HM. Cytochrome P450-catalysed pathways in human brain: metabolism meets pharmacology or Id drug with new mechanism of action? Pharmacol Ther 2007; 3:537-45.
84 Shukla R, Tyagi E, Kumar E. Protective effect of cox and nos inhibitors on Ips induced oxidative stress in rat. Annals of Neurosciences 2008; 15(1 ):6-10.
85 Lucas A, Moreley R, Lister G, et al. Effect of very low birth weight on cognitive abilities at school age. N Engl J Med 1992;326:202-03.
(c) Annals of Neurosciences.All Rights Reserved