Annals of Neurosciences, Vol 15, No 1 (2008)
Annals of Neurosciences, Volume 15, Issue 1 (January), 2008
MELATONIN OFFERS PROTECTION AGAINST GLUTAMATE RECEPTOR AGONISTS IN NEURONAL CULTURES
Corresponding Author:
Prof S Daya
Head of Pharmacology, Faculty of Pharmacy
Rhodes University, PO Box 94
Grahamstown, 6140
South Africa
Telephone: +27-46-603 8381
Fax: +27-46-6361205
E-mails.
(Date received: 14.09.07)
Abstract
The ability of melatonin to offer neuroprotection against glutamate, and the glutamate receptor agonists, kainate, N-methyl-D-aspartate and quinolinic acid was investigated using primary neuronal cultures. Exposure of neurons to each of the agonists for 20 minutes resulted in a significant reduction in cell viability as determined by trypan blue staining. Co-treatment with melatonin (0–500μM) and each of the agonists (420μM) resulted in a significant increase in cell viability. When melatonin (150μM) was added for 20 minutes prior to addition of any of the agonists (100μM), no protection occurred. Addition of a similar concentration of melatonin after a 20 minute incubation with various agonists, resulted in significant protection against all agonists. Simultaneous exposure to melatonin and agonist offered significant neuroprotection with all of the agonists except glutamate. The present report demonstrates that melatonin is able to offer neuroprotection against neurotoxicity induced by glutamate and the three glutamate receptor agonists.
Key Words : Melatonin, Glutamate, Kainate, Quinolinate, N-methyl-D-aspartate, Neuroprotection
Introduction
L-Glutamate is the most abundant free amino acid in the central nervous system existing uniformly in millimolar concentrations in the cytoplasm of most vertebrate neurons (1, 2). Apart from its role as a building block in the synthesis of proteins and peptides, glutamate is also a major neurotransmitter in the mammalian brain (3).
The role of glutamate as a neurotransmitter was only widely accepted in 1974, when Fonnum reviewed the data known at the time and showed that glutamate met all the criteria of a neurotransmitter (3). In the past decade, glutamate has also been shown to be a very potent neurotoxin. The concept of glutamate as an excitotoxin is largely due to the work of, JW Olney who demonstrated a correlation between the excitatory properties of various glutamate analogues and their ability to elicit neurotoxic damage (4).
Olney conceived the term “excitotoxin” to denote the group of excitatory amino acids (EAA's) which selectively killed neurons and cell bodies by depolarizing actions (5). With the development of various classes of glutamate receptor antagonists, it was demonstrated that excitotoxicity is a receptor mediated event, as antagonists can prevent both excitation and toxicity (4).
The mechanism by which excessive EAA receptor activation leads to neuronal death involves increased membrane permeability and abnormal Na+, Cl− and Ca2+ influx into the neuron. The first two ions disrupt the osmotic pressure of the neuron which can lead to lysis of the neuron.
Much of the toxicity associated with glutamate receptor activation is the result of excessive elevation of intracellular neuronal Ca2+ levels. There are many possible ways in which Ca2+ overload may kill cells, including activation of intracellular proteases and lipases, impaired mitochondrial function, and the generation of free radicals. Free radicals and products of free radical reactions can cause damage to macromolecular targets such as DNA, proteins, and cellular membranes. A particularly important consequence of free radical damage in cells, is the peroxidation of polyunsaturated fatty acids, which results in the formation of lipid peroxides and aldehydes (6). These products can cause extensive damage to membrane structure and integrity, and it is this damage that can result in death to neurons.
In the brain there are a number of different glutamate receptors that have very distinct pharmacological and physiological properties. EAA receptors have been characterised according to their effect on ionic balance (ionotropic) or secondary messenger systems (metabotropic). The ionotropic receptors are characterised as N-methyl-D-aspartate (NMDA), α-amino-3-hydroxy-5-methyl-4-isoxazole-propionic acid (AMPA), kainic acid (KA), and 2-amino-4-phosphonobutyrate receptors. Each of these receptors are coupled to ligand gated ion channels which are permeable to both Na+ and K+, and in the case of NMDA receptors, Ca2+ as well (1, 7).
In the present study we chose to work with glutamate and the glutamate agonists; KA, NMDA and Quinolinic acid (QA). NMDA is the agonist for the NMDA receptors which are the most populous of the glutamate receptors. Kainic acid is the neurotoxic analog of glutamate that interacts with the kainic acid receptor (8). When injected into animals, KA typically induces seizures and causes extensive neuronal damage. Melatonin has also been shown to protect against kainic acid induced toxicity in whole brain homogenate (9). QA acts at the NMDA receptor, but the toxicity that occurs is similar to that observed during kainate toxicity. QA is thought to be involved in Huntington's disease and concentrations of this neurotoxin are known to increase in the brain with age (10).
The neurohormone, melatonin, has been shown to be a very effective scavenger of hydroxyl-and peroxyl-radicals and has been reported to be effective at suppressing the accumulation of MDA and 4-HAD in brain homogenate (11, 12). Melatonin has been shown to be a more potent hydroxyl radical scavenger than glutathione, mannitol and vitamin E (12, 13). Melatonin is able to suppress the formation of cataracts in new born rats treated with the glutathione depleting agent, buthionine sulfoxine, and has also been shown to offer protection against δ-aminolevulinic acid-induced oxidative damage in the rat cerebellum (14, 15).
Melatonin has unique physiological properties which distinguish it from other antioxidants. In contrast to all other known low molecular weight antioxidants, the melatonin molecule, once oxidised, cannot be reduced or regenerated. This built in safety mechanism protects against auto-oxidative radical generation and toxic redox cycling (16).
It has been proposed that glutamate neurotoxicity enhances free-radical production, thereby causing neuronal death in a variety of age-related diseases. Using neuronal cell cultures, we decided to investigate the neuroprotective properties of melatonin against glutamate and three different neurotoxic glutamate agonists, namely NMDA, KA and QA.
Materials and Method
Glutamate, QA and melatonin were purchased from Sigma Chemical Company (USA). NMDA and KA were purchased from Research Biochemicals International (RBI) (USA).
Eagle's Minimum Essential Medium, Hank's Balanced Salts (modified form) and Cytosine-β-D-arabinofuranoside were purchased from Sigma Chemical Company. Trypsin and trypsin inhibitor were purchased from Boehringer Mannheim (Germany).
Foetal Calf Serum (FCS) was obtained from Delta Bioproducts (South Africa). All other chemicals were of the highest grade obtainable from commercial sources.
Primary Cultures
Primary neuronal cultures were established from the brains of day old rats. The brains from litter were pooled and minced into 1mm2 pieces. The brain tissue was then transferred to sterile 15ml tubes containing 5ml 0.2% trypsin in HBSS. After 20–30 minutes of incubation at 37°C in a shaking water bath, the tissue pieces were collected by centrifugation at 3500xg for 5 minutes at 4°C, rinsed with fresh HBSS, and incubated for a further 5 minutes at 37°C in soybean trypsin inhibitor. After another wash in HBSS, the cells were dissociated by titrating tissue through the narrow bore of a fire-polished pasteur pipette. Approximately 500 000 cells/ml were added to 3ml of MEM containing 10% FCS in 25cm2 culture flasks. Neurons were allowed to attach to the surface of the culture flask for 2½ hours, after which the media was discarded and replaced with 5ml MEM containing 10% FCS and 10μM cytosine-β-D-arabinofuranoside.
Agonist Exposure
All experiments were conducted in 7–10 day old cultures. The MEM was discarded from each flask and the neurons were washed with 3ml of HBSS. Thereafter the cultures, each containing 3ml of fresh MEM to which the agonists and/ or melatonin had been added, were incubated at 37°C for 20 minutes. Melatonin was dissolved in ethanol. Final ethanol concentration in cell culture flasks was 1.5%. The same concentration of ethanol was added to all control incubations. Following incubation, the cultures were washed twice with HBSS, before fresh MEM containing 10% FCS and 10μM cytosine-β-D-arabinofuranoside was added. The neurons were incubated for a further 18 hours at 37°C.
In order to determine when best to add melatonin in order to achieve maximum neuroprotection, melatonin was added either, before, during or after the incubation of the neurons with the agonist. Neurons treated with melatonin prior to addition of the agonist had sufficient melatonin added to the existing growth medium 20 minutes before the addition of the media containing the agonist. Cultures treated with melatonin during the agonist insult, had melatonin added to the incubation media, together with the agonist. Finally, in cultures treated with melatonin after the agonist insult, cultures were treated with agonist and melatonin was added to the fresh medium that was put into the flasks following the insult. Neurons were then incubated for 18 hours in the presence of melatonin.
Assessment of Neuron Viability
Trypan Blue staining was used to assess neuronal viability. Cultures were incubated in 10% of a 0.4% trypan blue solution for 5 minutes. Percent viability was assessed by counting the number of dead neurons which stain blue as well as the total number of neurons in five randomly chosen fields per plate using an inverted microscope.
Counting was done by an observer unaware of the experimental conditions.
Statistical Analysis
The results were analysed using a one-way analysis of variance (ANOVA). If the F values were significant, the Student-Newman-Keuls test was used to compare the treated and control groups. The level of significance was accepted at p<0.05.
Results
In all cases, treatment of the neuronal cultures with glutamate and the glutamate agonists; Kainic acid, NMDA and QA, resulted in death to a number of neurons. This was expressed as a decrease in cell viability in the culture medium as shown in Fig 1. QA was found to be the most neurotoxic of the agonists used. This is demonstrated by our finding that only 29.2% of neurons survived in flasks treated with 500μM of QA. Similar doses of Glutamate, KA and NMDA yielded survival rates of 38.7%, 44.1% and 54.3% respectively when compared to control cultures. Similar experiments conducted with melatonin up to a concentration of 1mM did not alter culture activity (results not shown).
The neuroprotective effects of co-treatment with melatonin and agonist (420gmM) showed that in all cases melatonin protects against the decrease in cell viability. This concentration was used as it most closely approximated the mean LD50 for the combined agonist experiments. As in Fig 2, the level of neuroprotection offered was dependent on the concentration of melatonin.
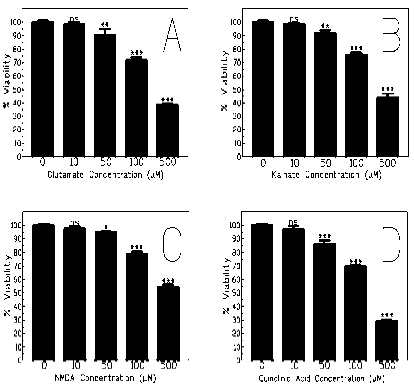
Figure 1: Effect on % viability of A) Glutamate, B) KA, C) NMDA, and D) QA. Primary neurons were incubated for 20 minutes with the various agonists, and % viability was assessed using trypan blue staining. Values represent the mean ± SEM (n=3–5) (*P<0.05; **P<0.01; ***P<0.001 in comparison to 0 controls).

Figure 2: Effect of melatonin on % viability when co-treated with A) Glutamate, B) KA, C) NMDA, and D) QA. Primary neurons were co-incubated for 20 minutes with Melatonin (0–500μM) and the agonist (420 μM). The % viability was assessed using trypan blue staining. Values represent the mean ± SEM (n=6–9) (*P<0.05; **P<0.01; ***P<0.001 in comparison to 0 controls).
Finally, we determined the optimal time to expose the cells to melatonin. A low dose of agonist (100μM) was used as this was the lowest dose of agonist that gave significant decrease in cell viability in the first set of experiments. A low concentration of melatonin (150μM) was also used so that excessive protection would not result. As shown in Fig 3, no protection was offered to neurons that were treated 20 minutes prior to agonist exposure. In flasks where cultures were co-treated with agonist and melatonin after some time, there was significant protection offered, except in the case of the flasks treated with glutamate, where no significant increase in cell viability was recorded in comparison to control cultures. Neurons treated with melatonin for 18 hours following the 20 minutes agonist exposure showed significant protection.
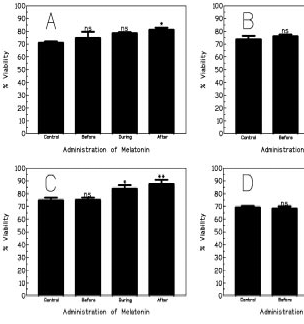
Figure 3: Effect of time of melatonin administration on % viability whe incubated with melatonin (150μM) either for 20 minutes before; dur the agonist incubation. The % viability was assessed using trypan b **P<0.01; ***P<0.001 in comparison to 0 controls).
Discussion
Two of the agonists, viz NMDA and QA, act on the NMDA receptor. When stimulated, this receptor allows Ca2+ to flow into the neuron. Over stimulation can thus lead to excessive Ca2+ influx (17). Calcium has been shown to stimulate a number of pathways that can give rise to the production of free radicals. Various agents that reduce intracellular calcium concentrations following an insult from an NMDA agonist have been shown to offer neuroprotection. Melatonin has not been shown to reduce Ca2+ levels in neurons, but it is a very powerful antioxidant. We therefore propose that melatonin offers its protection by removing free radicals that are produced by pathways stimulated by Ca2+.
In contrast to the report by Giusti et al., melatonin was not able to protect against NMDA induced neurotoxicity (18,19). We however found that melatonin offers neuroprotection against NMDA, and QA which acts at the NMDA receptor. Reasons for the discrepancy could be due to different experimental protocols. This could be due to the fact that the neurons were only exposed to agonist for 20 minutes, while the previous authors had exposed their neurons to NMDA for 60 minutes. The primary neuronal cultures also came from neonates of different ages. We would propose that an increase in the intracellular Ca2+ concentration brought about the over activation of the NMDA receptor, results in the over production of superoxide anions. Because of the shorter incubation times under our experimental conditions, there would not be the same rise of superoxide anions. Other free radicals produced would be effectively removed by the melatonin, so providing neuroprotection.
Our results further show that melatonin acts as an antioxidant due to the fact that when administered before the insult, melatonin was not able to offer any protection. This is to be expected as there would only be an increase in superoxide anion production following the calcium influx brought on by the agonist insult. Apart from neuronal cultures treated with melatonin during glutamate incubation, in all other cultures that were treated with melatonin at the same time, or after the insult, neuroprotection was offered. Although Giusti et al found that melatonin did not offer protection when administered in the 18 hours following treatment by KA, our findings would reinforce the argument that melatonin is providing protection as an antioxidant. The presence of melatonin at the same time as the insult would allow melatonin to enter the neuron and remove free radicals as these were produced. The production of free radicals occurs as a result of an increase in intracellular calcium, so following the insult, intracellular calcium levels would still be elevated within the neuron. Melatonin should therefore still be effective at removing free radicals produced even after the insult has been removed.
The present report shows that melatonin is able to offer neuroprotection against glutamate, and the glutamate agonists; KA, QA and NMDA. Melatonin must be administered during or after the agonist insult in order to offer protection. It appears that the melatonin offers protection by acting as an antioxidant, thus removing dangerous free radicals from the neurons. Melatonin levels are known to decrease throughout life, such that in old animals, including humans, blood and tissue levels of melatonin are low. Glutamate neurotoxicity is believed to play a part in many age related neurodegenerative diseases, including Alzheimer's disease, while Huntington's disease is associated with elevated QA levels in the brain. These results demonstrate that there is definitely a role for melatonin in the antioxidant defence system of an organism.
Acknowledgement
This work was sponsored by a grant to Prof S Daya from the Medical Research Council of South Africa.
References
1. Greenamyre JT, Porter RHP. Anatomy and physiology of glutamate in the CNS. Neurology 1994; 44: 7–13.
2. Tilson HA, Mundy WR. Basis of excitatory amino acid system-related neurotoxicity. In: Chang LW, Slikker W Jr (Eds) Neurotoxicology: Approaches and methods, Academic Press, London, 1995; 359–70.
3. Fonnum F. Glutamate: A neurotransmitter in mammalian brain. J Neurochem1984; 42: 1–11.
4. Olney JW. Excitatory amino acids and neuropsychiatric disorders. Biol Physiology 1989; 26: 505–25.
5. Coyle JT, Puttfarcken P. Oxidative stress, glutamate, and neurodegenerative disorders Science 1993; 262: 689–95.
6. Ottino P, Duncan JR. Effect of α-tocopherol succinate on free radical and lipid peroxidation levels in BL6 melanoma cells. Free Rad Biol Med 1997; 22: 1145–51.
7. MacDonald JF, Nowak LM. Mechanisms of blockade of excitatory amino acid receptor channels. Trends Pharmacol Sci 1990; 11: 167–171.
8. Rose CR, Ransom BR. Mechanisms of H
9. Meldrum B, Garthwaite J. Excitatory amino acid neurotoxicity and neurodegenerative disease. Trends Pharmacol Sci 1990; 11: 379–84.
10. Moroni F, Lombardi G, Moneti G, Aldinio C. The excitotoxin, quinolinic acid is present in the brain of several mammals and its cortical content increases during the ageing process. Neurosci Let 1984; 47: 51–5.
11. Tan DX, Chen LD, Poeggeler B, Manchester LC, Reiter RJ. Melatonin: A potent, endogenous hydroxyl radical scavenger. Endocr J 1993; 1: 57–60.
12. Pieri C, Marra M, Moroni F, Recchioni R, Marcheselli F. Melatonin: A peroxyl radical scavenger more effective than vitamin E. Life Sci 1994; 55: 271–6.
13. Melchiorri D, Reiter RJ, Chen LD, Sewerynek E, Nistico G. Melatonin affords protection against kainate-induced in vitro lipid peroxidation in brain. Eur J Pharm 1996; 305: 239–42.
14. Abe M, Reiter RJ, Orhii P, Hara M, Poeggeler B, Barlow-Waldon LR. Inhibitory effect of melatonin on cataract formation in newborn rats: Evidence for an antioxidant effect for melatonin. J Pineal Res 1994; 17: 94–100.
15. Princ FG, Maxit AG, Cardalda C, Batlle A, Juknat AA. In vivo protection by melatonin against delta-aminolevulinic acid-induced oxidative damage and its antioxidant effect on the activity of haem enzymes. J Pineal Res 1998; 24:1–8.
16. Poeggeler B, Reiter RJ, Ta n DX, Chen LD, Manchester LC. Melatonin, hydroxyl radical-mediated oxidative damage, and ageing: A hypothesis. J Pineal Res 1993; 14: 151–68.
17. Lu YM, Yin HZ, Chiang J, Weiss JH. Ca
18. Giusti P, Lipariti M, Franceschini D, et al. Neuroprotection from melatonin from kainate-induced excitotoxicity in rats. FASEB J 1996; 305: 239–42.
19. Giusti P, Gusella M, Lipariti M, et al. Melatonin protects primary cultures of cerebellar granule neurons from kainate but not from N-methyl-D-aspartate excitotoxicity. Exp Neurol 1995; 131: 39–46.
(c) Annals of Neurosciences.All Rights Reserved