Annals of Neurosciences, Vol 12, No 3 (2005)
Annals of Neurosciences, Volume 12, Issue 3 (July), 2005
NEUROLOGICAL SYNDROMES AT HIGH ALTITUTDE PART-II
PVS Rana
Manipal College of Medical Sciences, Pokhara Nepal
Corresponding Author Dr. PVS Rana
Prof. Medicine and Neurology,
Director Clinical Program,
Manipal College of Medical Sciences, Pokhara Nepal
Email
Tel: 0977-61-526416-196
Abstract
Impairment higher mental functions and impaired decision making which develops at the altitude of 3080 meters and above, further add to the high mortality associated with high altitude illnesses. In hospital based studies of ischemic stroke forms 13.7/1000 of hospital admissions from high altitude areas as compared to 1.05/1000 in non high altitude areas and accounting for 21.3 – 35% of cases. Anoxia induced capillary damage, hematological alteration leading to increased viscosity and development of hypercoagulable state are suggested as the cause of ischemic strokes. A favorable good prognosis is noted in these cases with antiplatelet aggregating agents, and on evacuation to lower altitude. Hemorrhagic stroke also noted due to multiple factors including anoxia induce capillary damage, increase cerebral blood undue physical exertion at high altitude flow and development of high altitude cerebral edema. Available literature on the neuropathy at high altitude is reviewed. Twenty five cases of idiopathic intracranial hypertension developing at high altitude is reported and discussed in detail. This condition is not reported so far in literature. Detailed investigations are needed to exclude other causes and sinovenous thrombosis. The latter requires anticoagulant therapy in addition to other measures. The prognosis in these case is favorable with early recognition, early treatment and on evacuation to lower altitude. The neuropathy cases can be prevented with adequate health education and proper planning before induction to high altitude. The persons who shad suffered fro stroke should avoid induction to high altitudes.
Key words:
High altitude, Acute mountain sickness (AMS), High altitude cerebral edema (HACE), High altitude pulmonary edema (HAPE), Ischemic cerebrovascular accident (ICVA), Hemorrhagic strokes, Acclimatization, Neuropathy, Trench Foot, Frost Bite, Papilloedema, Idiopathic intracranial hypertension.
Introduction
Human desire to conquer the mountains has led to development of many sport activities and expeditions at high altitude. These activities are entertaining, thrilling and exciting but are dangerous due to natural disasters lurking at high altitudes. In addition, induction to high altitude exposes the inductee to ill effects of increasing hypoxia. Brain bears the main brunt of decreased oxygenation at high altitudes. The effect may be fatal if anoxia is acute and severe. While acute decrease in arterial oxygen saturation to 85% impair mental concentration, fine motor coordination, fall of oxygen saturation to 75% results in poor judgment, irritability and decreased motor functions. Difficulty in learning new tasks is noted at 3048 meters. Sensory, motor and perceptual performance decreases at 6100m (1, 2). There is electroencephalographs (EEG) evidence of cortical suppression in fresh inductee at 3050 meters as compared to native's (3). The type and rate of EEG abnormality was related to symptoms of irritability, insomnia and depression in healthy subjects at 4100m (4). High altitude headache (HAH), acute mountain sickness (AMS), high altitude cerebral edema (HACE) and high altitude pulmonary edema (HAPE) grouped under "High Altitude Illnesses", are the commonest neurological syndrome produced at high altitude and are extensively studied and reviewed. Present communication is an attempt to highlight the other neurological syndromes at high altitude (5) with special reference to cerebrovascular accidents, syndrome of idiopathic intracranial hypertension and neuropathies mostly mentioned as case reports only.
Cerebrovascular Accident at High Altitude
(a) Ischemic cerebrovascular accidents at high altitude
Cerebral ischemic episodes are expected at high altitude due to increasing hypoxia. There is limited knowledge about stroke at high altitude and mostly reports are of isolated cases of focal ischemic neurological syndromes. Hackett et al., 1987(6) reported 7 cases of cortical blindness (i.e. symptoms of intermittent blindness with normal pupillary reflex) in high altitude climbers and trekkers. Transient expressive aphasia, in an acclimatized high altitude climber, was reported by Deitz & Mckeil, 2000 (7) and was thought to represent a migranous aura. Litz & Bishop, 2000 (8) reported a case of transient global amnesia (TGA) at high altitude. There are only few detailed studies on strokes at high altitude (9–14). Singh and Chauhan, 1972 (9) reported clinically evident venous thrombosis in peripheral and splanchnic veins and arterial thrombosis in coronary, mesenteric and cerebral vessels in troops, weeks after their arrival at high altitudes. They suggested the role of increased platelet adhesiveness in thrombus formation. While studying ischemic cerebrovascular accidents (ICVA) in young soldiers, Vijayan et al., 1974 (10) noted that 35% had onset of illness at high altitude (3500–5000 meters). These patients had no other high altitude related illnesses, polycythemia, raised hematocrit or evident risk factors in them.
Rana et al., 1978 (10) investigated 108 cases of ischemic strokes in young soldiers, below 45 years of age. Twenty three (21.3%) patients had onset at high altitude while serving above 3500 meters. One (4.3%) patient gave history of HAPE while another patient, a local resident of high altitude area, had pulmonary hypertension. One (4.3%) patient had ischemic stroke in the past from he had recovered fully. Thirteen (56.5%) patients were smokers. No evident source of embolism was detected. There was no relation to duration of stay or number of induction to high altitude. None had other features of other high altitude illness at the onset of stroke. As a group their hemoglobin range from 14–15 gm/dl (mean 14.8.gm/dl) and hematocrit value were 36–49%. Platelet adhesiveness and plasma fibrinogen was increased while fibrinolytic activity was reduced suggesting a hypercoagulable state and was presumed to be the cause as all patients were young and lacked risk factors except smoking and stay at high altitude. All the patients were evacuated to plains and treated with asprin 300 mg twice a day. Six (26.08%) cases angiographic abnormalities which included proximal internal carotid artery (ICA) block in 2(8.6%), middle cerebral artery (MCA) block in 2(8.6%), hypoplasia of ICA and tortuosity of intracranial vessels (Figure 1 & 2) in one (4.3%) case each respectively. Initially development of tortuosity was thought akin to the development of tortuosity of ophthalmic arteries at high altitude. However, when angiographic abnormalities in 108 cases were reviewed this abnormality was noted in 11.1% of cases (15). Follow up 108 cases (16) revealed a mortality rate of 4.4%. While 63% were independent, 10.1% needed supervision only, 32.2% needed assistance and 2.2% were dependant for acts of daily living respectively. All the 23 patients with onset of stroke at high altitude recovered and could be rehabilated back to their parent jobs. The patient with tortuosity of intracranial vessels (Figure-1) and 4 years later presented as CP angle tumor (Figure-2).
Song et al., 1986 (12) reported four cases of cerebrovascular accidents in subjects who climbed above 5000 meters. Except in one, who developed stroke on second day, in other it developed 3 weeks after the induction to high altitude. Unlike cases of arterial thrombosis reported earlier, these patients had cerebral venous thrombosis. Increased viscosity, secondary to raised hematocrit and dehydration consequent to less fluid intake and increased loss resulting from rapid breathing in low humid air and increased sweating was suggested as the cause as all patients had polycythemia and raised hematocrit detected in 2 patients. That cerebral venous thrombosis occurs at high altitude was shown in their autopsy study by Dickinson et al., 1983 (13).
Jha et al., 2002 (14) presented the clinical profile of 30 cases of stroke at high altitude seen Command Hospital (WC) Chandimand ir between November 1998 to July 2000. Strokes formed 13.7/1000 of hospital admissions from high altitude area, compared to 1.05/1000 in non high altitude area. All cases from high altitude area were males (serving soldiers of armed forces). Their mean high altitude stay was 10.2 months, and they were all located at heights greater than 4270 meters. Their age ranged from 22 to 48 years (mean 33.4 yr). All except 2 were below 45 years of age. Except for smoking (in four cases), they had no preexisting risk factors. Twenty-six cases were of ischemic stroke which included four cases of transient ischemic attacks and reversible ischemic neurological deficits, 2 of intracerebral hemorrhage, and 2 had cerebral venous thrombosis. Polycythemia with Hb ranging from 16.2 to 22 g/dl was seen in 21 of these 28 cases (75%). CT scan showed massive infarcts involving at least 50% of one cerebral hemisphere in 12 cases. Multiple infarcts were seen in one case. These workers stressed on the role of polycythemia as important risk factors. None had positive antiphospholipoid antibody. Number of cases was few to asses the role of Protein C & S deficiency which was detected in one case each.
In post CT scan era (present communication) the author investigated 51 high altitudes related neurological syndromes between the year 1990–2004 in defense services hospitals and at Manipal Teaching Hospital, Pokhara (Nepal). These included ischemic strokes-25 cases, hemorrhagic stroke-11 cases (i.e. intracerebral hemorrhage-7; subarachnoid hemorrhage-3; subarachnoid and intracerebral hemorrhage-1) and 15 cases presented as raised intracranial tension without localization. The ischemic strokes cases included 15 soldiers, aged 25–45 years, who were deployed at high altitude (above 3500 meters) for 5–18 months and 10 civilians ( age group 30–50 years) residing or trekking at a height between 2500–4500 meters. The clinical picture corresponded to the vascular territory involved. While 22 cases presented as carotid artery stroke (i.e. large vessel stroke 20: small vessel stroke- 4), vertebrobasilar stroke was noted in 3 cases (upper lateral pontine syndrome 1: Lower lateral pontine syndrome-1: brain stem and cerebellar-1) and spinal stroke in one case respectively. Short summary of four cases is given below:
Case 1: A 35 years old Irish lady developed acute mountain sickness and spinal stroke with clinical picture resembling syndrome of artery of Adamkiewicz (great radicular artery) syndrome while she was trekking at the height between 2500–3500 meters. She was treated with steroids and aspirin therapy. She was making good neurological recovery when she returned to her native country and was lost for follow up.
Case 2: A 28 years lady from UK developed right focal seizure with generalization followed by development of right hemiplegia and classical picture of opercular syndrome. She gave history of left MCA territory stroke 2 years back from which she had made full recovery and was on antiplatelet therapy. She developed above symptoms while trekking at a height between 3000–4000 meters. The CT scan showed two areas of infarcts in opercular areas. She made rapid motor recovery but her dysphagia and expressive dysphasia persisted. She returned to her country and lost for follow up.
Case 3: A 35 years old trekker and guide developed acute mountain sickness and right lower lateral pontine syndrome. He had polycythemia. The CT scan, MRI angiography and venography were normal. With steroid and aspirin therapy he made good neurological recovery. Only symptom of paraesthesia on right side is persisting. He was advised to change his profession.
Case 4: A 23 years old male serving in high altitude developed HACE and HAPE On examination he was cyanosed neurological examination revealed disorientation, slurred speech, right hemiparesis and bilateral extensor plantar response. He had polycythemia. The CT scan revealed periventricular luscencies. The ECHO study revealed transient thrombus in right ventricle.. MRI could not be done. With diuretics, steroids, aspirin and other supportive measures he made full recovery.
When followed up after 2–6 months all patients were independent for act of daily living, which was consistent with our earlierobservation. The comparison of the clinical profile of these cases with those of Jha et al., (2002) is shown in Table-2.
Hypoxia produces hematological changes. However, studies on coagulation parameters on individual having strokes are limited. Singh et al., 1969 (17) reported increased fibrinogen levels and prolonged clot lysis times in HAPE patients, which they attributed to breakdown of fibrinolytic system. Singh et al., 1974 (18) also reported thrombotic occlusive of pulmonary vessels in hypertensive pulmonary vascular disease in high altitude pulmonary hypertension cases and presumed the similar mechanisms responsible for ischemic strokes as well. Rana et al., 1975 (11) reported increased platelets adhesiveness, increased fibrinogen levels and decreased fibrinolytic activity in their studies of ischemic strokes which included cases having onset at high altitude. These findings suggested a role of hypercoagulability in production of ischemic stroke at high altitude. In addition, hypoxia produces (a) hyperventilation induced cerebral vasoconstriction which partially offset the hypoxia induced increase in cerebral blood flow (CBF). CBF flow was found to increase 33% after 12 hours of induction at 3800m in spite of fall in PCO2 from 40 to 35%. After 4–5 days CBF decreased but was still 14% greater than at sea levels (19); (b) secondary polycytmeia through hypoxia induced increased erhythropoietin; (c) dehydration through combined effects of increased mechanical work of hyperventilation, increased physical activity, breathing through humid air, decreased thirst and ultraviolet rays at high altitude and aggravated further by alcohol intake, use of diuretics and The hemoconcentration thus produced raises the serum viscosity which impairs the cerebral blood flow. In addition hypoxia induces capillary damage may promotes intravascular thrombosis. Jha et al., 2002 (14) found low levels of Protein C & S but its role is not clear due to study in large number of cases.
Hemorrhagic strokes at high altitude
Though ischemic strokes are commonly seen, hemorrhagic strokes are also noted at high altitude. Dickinson et al., 1983 (13) in a autopsy study of seven trekkers, who died during expedition reported hemorrhagic infarcts associated with cerebral edema in cortex, sub cortical white matter of cerebrum and brain stem (case-3), smaller hemorrhages and petichae in subcortical white matter of cerebrum, corpus callosum, pons and cerebellum (case-5), large hemorrhage in parietal lobe (case-2) and subarachnoid hemorrhage in 2 cases respectively. In none aneurysm or AV malformation was detected. In this study all patients had cerebral edema while pulmonary edema was present in 4 cases only. Ophthalmologic findings included tortuosity of blood vessels in all cases, papilloedema in 5 cases and hemorrhage in 4 cases respectively. Venkatraman et al., 1993 (20) reported 5 cases of hemorrhagic stroke (i.e. bilateral cerebral hemorrhage-1, parietooccipital hemorrhage-1, deep seated hemorrhage-2, Subarachnoid hemorrhage (SAH)-2 and SAH with ICH-1), developing at high altitude. None had any evidence of bleeding disorders or aneurysm or vascular malformation. Jha et al., 2002 (14) in their study reported 2 patients of hemorrhagic strokes having onset at high altitude. The author (present communication) detected 7 cases of hemorrhagic stroke (i.e. intracerebral-5; basal ganglion hemorrhage-2) 3 had subarachnoid hemorrhage and one case had booth subarachnoid and ICH. Detailed investigations revealed no symptomatic cause. There is no report to suggest increased bleed from aneurysm or from AV malformation at high altitude which can be expected as a result severe exertion and increased cerebral blood flow and volume in early stages of induction. Role of increased cereal blood flow, development of HACE and hypoxic capillary damage appears to be contributing factors in these cases (13).
2. SYNDROME OF IDIOPATHIC INTRACRANIAL HYPERTENSION AT HIGH ALTITUDE:
Induction to high altitude results in acute cerebral edema, which can be associated with frank papilloedema (13,17). Such patients need detailed investigations as other diseases associated with raised intracranial tension can also manifest at high altitude. Papilloedema due to chronic raised intracranial tension was an interesting entity reported in Indian soldiers serving at high altitude (20). Additional fifteen patients with similar clinical picture were detected during this period (present communication). All, except two patients, were in soldiers serving at the height of 3500–5500 meters with duration of stay ranging from 6–18 months. The age group varied from 25–45 years. Of the two civilian cases, one was residing at a height of 2300 meters with frequent visit to higher altitude and the other was working as porter during trekking between 3000–4000 meters. All presented with chronic headache and had frank papilloedema without localizing signs. The only other symptom of atypical absence attack was noted in one case. Retinal vessels were dilated and tortuous. Two patients had hemorrhages as well. None had visual field defects. Polycythemia was noted in 6 patients with Hb ranging from 14.5–17 Gm/dl. Metabolic parameters and CSF analysis were normal. CT scan revealed no intracranial space occupying pathology or classical signs of cortical venous thrombosis (CVT).The ventricle size were normal. In two patients abnormal enhancement of transverse sinus was noted. Magnetic resonance imaging venography (MRIV) revealed sinus thrombosis in the case with atypical absence attack (Figure-3) along with signal void in lateral sinus. All patients made full recovery with anti-cerebral edema measures (i.e. acetazolamide/frusemide) and on evacuation to sea level. The case having CVT was treated with anticoagulant therapy.
Marked changes are noted in retinal circulation on rapid induction to high altitude areas. These includes increase in retinal vessel's diameter, tortuosity of retinal vessels, increase in retinal blood flow and volume with diffuse punctuate or confluent flame shaped hemorrhages of variable size near the disc and sparing macula. Frank papilloedema may develop. Rapid increase in intracranial tension causes effusion of CSF into optic disc leading to compression of ophthalmic vein and opening of choroido-retinal anastomotic channels causing venous hypertension and retinal hemorrhage (21). Flame hemorrhages are most commonly seen but cotton wool spots, dot and blot, and pre-retinal hemorrhages have also been reported (22). They are symptomatic only if involving the macula and majority of hemorrhage resolve within 7 to 14 days of descent. Severe physical exertion may also cause retinal hemorrhage by decreasing intraocular pressure and transmission of systemic pressure to already embarrassed retinal circulation. With decent to low levels these hemorrhages get absorbed spontaneously. These acute changes characteristics of high altitude retinopathy (a pathological response by the retina to high altitude hypoxia) occurs in up to half of those ascending above 2500 meters (22). Unlike acute retinopathy, in present cases development of raised ICT & papilloedema was gradual. Dilated and tortuous retinal vessels noted in fundus of patients having chronic mountain sickness (CMS). However, CMS has not been reported in local population and in soldiers living in high altitude areas up to 42 months (17).
The clinical profile was that of idiopathic intracranial hypertension (IIH) with the difference that as they had onset at high altitude. The syndrome of intracranial tension without hydrocephalus, intracranial neoplasm and normal cerebrospinal fluid (CSF) is a recognized clinical entity and was named as "Benign intracranial hypertension" by Foley, 1955 (23). Later, an alternative terminology of idiopathic intracranial hypertension (IIH) was recommended for such cases (24). The cases having intracranial sinus venous thrombosis should be excluded (25), as it is a common mode of presentation of limited sinovenous thrombosis involving lateral sinus. In a careful study of 17 cases of proved sinus thrombosis on MRI venography or digital subtraction angiography (DSA) and 27 cases where sinus thrombosis was excluded, no symptoms or sign differentiated the group (25). Non specific finding neuro imaging include dilated cisterna magna and optic nerve sheath (26), empty sella, flattening of posterior sclera, tortuosity of optic nerve (27), giant arachnoid granulation and signal gap in lateral and sigmoid sinus (28). The pathogenesis of the syndrome is still poorly understood. Presently favored mechanism is increased resistance to CSF outflow at arachnoid villi, either due to primary disturbance in the CSF absorption mechanism at arachnoid villi or secondary to venous hypertension. A 3–5 folds decrease of CSF absorption (29) and raised pressure in venous sinuses is reported in IIH (30). How CSF absorption is affected is not clear. Delayed maturation or fewer arachnoid villi may be the primary cause or it may be secondary to compression of the arachnoid villi and the intracranial venous sinus due to raised intracranial tension from any cause (31). In a recent study the pressure gradient, demonstrated in transverse sinus, disappeared after CSF drainage (32). Similarity between pulmonary hypertension and chronic raised ICT is worth speculating. Like thrombosis in pulmonary microcirculation in cases of pulmonary hypertension, thrombosis in retinal microcirculation and in sinus venous systems may result in outflow obstruction and raised ICT. The recognition of this entity is important. However, the diagnosis should be made after exclusion of other causes of raised intracranial tension which can manifest at high altitude secondary to changes in cerebral blood flow and development of cerebral edema associated with other high altitude illness. Treatment of this disorder depends upon the presence or absence of visual symptoms and visual fields defects and should be graded (24). Various modalities of treatment includes repeated lumbar puncture, diuretics, steroids, shunting procedures and optic nerve fenestration but none has been studied by randomized controlled trial. In addition to evacuation to lower altitudes, similar treatment is to be followed in the cases having onset at high altitude. In those having CVT anticoagulant therapy may have a role.
Neuropathy at high altitude
Neuropathy can be expected at high altitude as in addition to alteration in sensations, exposure to cold adversely effects the nerve conduction. Conduction velocity is reduced by 1–2% of normal for myelinated fibers and 4–5% for unmyelinated fibers respectively on exposure to cold.). The conduction fails earlier and recovers less rapidly in smaller than in larger myelinated fibers. Development of neuropathy in experimental animals was first demonstrated by Deny Brown et al., 1945 (33). They studied cat sciatic nerve frozen with carbon dioxide snow and reported development of neuropathy with sparing of vessels and connective tissue cells. Seigmund, 1967 (34)) noted axonal degeneration with secondary affection of sympathetic ganglion and spinal cord due to cold induced circulatory and permeabilty changes. Neuronal damage occurs only when animals are exposed to temperature below 200 C and affect both large and small fibers with severity odamage proportional to lowering of temperature. Immediately after cold injury conduction block, cessation of axonal flow and axonal swelling occurs. Both myelinated and demyelinated fibers are affected. It is followed by axonal atrophy and frank degeneration affecting large myelinated fibers. Unmyelinated axons were spared (35).
(a) Neuropathy associated with Trench Foot
Trench foot is caused by exposure to cold at non freezing temperatures. Most of the clinical data on Trench food is based on the experience of soldiers exposed to non freezing cold injury during wars. In Falkland War 20% of the soldiers had non-freezing cold injuries while many other failed to report sick (36). Its clinical features includes (a) Exposure stage or stage of Chill blain during which the limb feel numb and powerless develops by swelling and the changes in its skin color, (b) Prehyperaemic stage characterized by numbness of limb, distal sensory loss of all modalities and distal muscular weakness or paralysis. All pulsations are absent in the affected limb which is swollen, cold, pale or blue in color, (c) Hyperaemic stage developing 2–4 hours after the exposure. The affected limb is hot, red, and has normal pulsations. This is associated with severe burning pain and paresthesias reaching to maximal intensity after 24–36 hours. Later, the degree of swelling increases with blister formation and echymosis especially if limb is rewarmed rapidly: Tissue necrosis or gangrene formation: may occur. With the development of hyperaemic state, there is partial improvement in sensation with marked hyperesthesia over the kin where sensation returns. However, distal parenthesis and muscular weakness with muscle wasting and clawing of toes may persist in some cases. This stage usually last for 6 to 10 weeks ranging from few days to several months and (d) Post hyperemic stage: during this stage the recovery continues. While mild cases recover fully, residual deficits persist in severely affected cases. The limb becomes cold with episodic feeling of warmth and hyperhydrosis at the margins of anhydrotic and analgesic areas. The sensory symptoms of persistent pain and paraesthesia along with distal sensory loss may persist with electrophysiological evidence of neuropathy i.e. prolonged distal latency absent sensory potentials and diminished sensory action potentials in sural nerve in the affected limb as late as six months after the exposure.
(b) Neuropathy associated with Frost bite
Frost bite results on exposure to sub zero temperatures which results in freezing of extra cellular fluid with ice crystal formation, inhibition of intracellular enzyme system and ischemic injury Latter, resulting in capillary damage, exudation of plasma, intravascular sludging and local thrombus formation, opening of AV shunts by passing the affected part, and subsequent development of gangrene. Only few studies on neuropathy associated with frost bite are reported in humans. In a detail study of neurological manifestations in 173 cases of frost bite, Suri et al.,1968 (37) reported symptoms of numbness in 76%, tingling in 72%, electric shock like sensation in 50.3%, burning dysaesthesia in 69% respectively. Sensory loss beyond the affected part was noted in 74% patients. Late autonomic changes in the form of electric like sensations, sweating abnormalities and skin atrophy was also noted in this study and was thought to be due to changes in posterior root ganglion as reported in earlier experimental studies (38), Electrophysiological studies were conducted after 3–5 weeks of the onset of frost bite. It revealed a delayed conduction velocity in 25% of cases while the distal latency was prolonged in 66.6% of patients respectively. These findings pointed toward a demyelinating pathology as reported earlier (34, 39). Similar findings were reported by Dash, 1974 (40) in subjects of frost bite after 3–4 weeks. In this study sural nerve biopsy showed evidence of demyelination.
(c) Neuropathy at high altitude unrelated to Trench Foot or Frost bite.
(i) Sensation of tingling paraesthesia and pain is produced when hand is immersed in cold water at 15–160 C for 1–2 minutes. While paraesthesia disappears, the pain fluctuates in severity before changing to unpleasant deep seated ache. The severity of pain increasing with further lowering of temperature. Immersion in very cold water (below 40 C) produces severe burning sensations in skin of the hand (Skin pain) which was thought to be arising from cutaneous thermoreceptor (41). Microneurography studies in man have shown that exposure to cold produces receptor desensitization with development of anaesthesia. (42). Mesones etal, 2002 (43) reported, an unusual syndrome of neuropathic pain and/or dysesthesia in both feet apparently unrelated to frostbite or trench foot injuries in 8 (4.8%) of 165 European mountaineers during 19 expeditions to the Himalayas in the years 1977 to 2000. Mountaineers complained of persistent and continuous pain, which was consistently described as a "corky" sensation in their feet, associated with severe lancinating exacerbations. Pain improved with cold and worsened with heat and gentle pressure. Symptoms were incapacitating in a third of the cases. Treatment with carbamazepine was effective, and the disorder evolved to total resolution in 4 to 8 weeks. Complete neurological work up of one such case after 14 days after its presentation, were unrevealing. The paucity of information regarding this particular variety of neuropathic pain of the feet may be due to lack of clinical suspicion in the field, favorable outcome.
(ii) Neuropathy in chronic mountain sickness: Chronic mountain sickness (CMS), a condition usually seen in acclimatized natives and at time also noted in fresh inductee who shows no sign of acclimatization or have hypoxic pulmonary vascular disease (i.e. secondary CMS). The neuropsychiatrie symptom dominates the clinical picture in addition to right sided heart failure secondary to pulmonary hypertension. Paraesthesia affecting limbs is a common symptom in them. Thomas et al., 2000 (44) reported mild sensory neuropathy manifesting with burning feet burning hand syndrome in 10 cases chronic mountain sickness and in four out of five healthy controls residing at 4338 meters altitude. Sural nerve biopsy, done in 3 cases, revealed Demyelinating neuropathy with reduction in unmyelinated axon population, and reduced thickness of basal lamina zone in endoneural vessels. The symptoms resolve centrifugally on descent to low altitude. This suggested altered axonal transport as the possible etiological mechanism. The reduced thickness of basal lamina zone pointed towards adaptive structural changes to hypobaric hypoxia similar to those occurring in other tissues.
| Parameters | Jha et al. (2002) | Rana (2005>) |
|---|---|---|
| Number of cases | 28 | 25 |
| Age (years) | 25–48 | 25–50 |
| Altitude (meters) | above 4700 | 2500–4500 |
| Civilian | ----------- | 15 |
| Smoking | 4(14.3%) | 8 (32%) |
| Hb > 16Gm/dl | 21(75%) | 7 (28%) |
| Protein C deficiency | 1 | ------- |
| Protein S deficiency | 1 | 2* |
| CT MRI | ||
| Abnormal | 24(80%) | 21(84%) |
| Infarcts | 22 | 20 |
| CVT | 2 (7.1%) | 1 (4%) *Estimated in 7 cases only. |
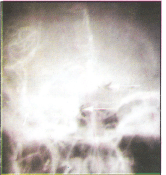
Figure-1: Carotid angiogram showing marked toruosity of intracranial vessels
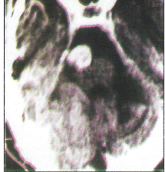
Figure-2: CT scan of the same patient done after 4 years showing tortuous vessels presenting as cerebello-pontine angle (CPA) tumor
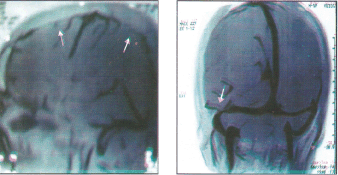
Figure-3: Idiopathic intracranial hypertension: MRI Venography showing (a) cortical venous sinus thrombosis and (b) signal void in lateral sinus.
Conclusions
Neurological syndrome of strokes, syndrome of idiopathic intracranial hypertension and neuropathies at high altitude are reviewed. A good recovery is noted in both ischemic hemorrhagic stroke cases on evacuation to low altitude. Aspirin therapy is the mainstay of treatment of ischemic stroke. Steroids have a role in those cases having AMS or HACE. Syndrome of idiopathic intracranial hypertension also recovers on evacuation to low altitude. However, these cases need investigations to exclude cortical venous thrombosis where anticoagulants are the mainstay of treatment. Neuropathies may develop at high altitude. The neuropathies associated with trench foot and frostbite may be prevented. Those inducted should have proper physical checkup and those having risk factors of stroke or have suffered it earlier should not be allowed induction to high altitude.
References
1. Cahoon PL. Simple decision making at high altitude. Ergonomics, 1972; 14: 157–164.
2. Kobrick Jl. Effects of hypoxia on peripheral visual response to dim stimuli. Percept Mot Skills 1975; 41: 467–464.
3. Selvamurthy W, Saxena RK, Krishnamurthy N, et al. Changes in EEG pattern during acclimatization to high altitude (3500meters) in man. Aviat Space Environ Med 1968; 49: 968–971.
4. Forster HV, Soto RJ, Demsey JA, et al. Effect of sojourn at 4300 meters altitude on electroencephalogram and visual evoked response. J Appl Physiol 1975; 39; 109–113.
5. Rana PVS. Neurological complications at high altitude. In Murthy JMK (Ed) Reviews in Neurology, Mundrika Graphics Hyderabad (India), 1994, Vol 1, pp 67–78.
6. Hackett PH, Hollingshead KF, Roach R, et al. Cortical blindness in high altitude climber and trekkers-A report of seven cases (abstract) In: Sutton JR, Houston CS, Coates G (eds). Hypoxia and cold. New York Praeger Press, 1987,536.
7. Dietz TE, McKiel VH. Transient high altitude expressive aphasia. High Alt Med Biol. 2000; 1:207–11.
8. Litch JA, Bishop RA.High-altitude global amnesia.Wilderness Environ Med 2000; 11:25–28.
9. Singh I, Chauhan IS. Abnormalities of blood coagulation at high altitude. Int J Biometeorol, 1972; 16:283–297.
10. Vijayan GP, Suri ML, Pratapa Rao WS, et al: Stroke in young in Armed Forces. AFMRC Project 529/73.
11. Rana PVS, Suri ML, Pratapa Rao WS, et al: Study of cerebra vascular disease in young in Armed Forces with special reference to treatment, AFMRC Project 708/75.
12. Song SY, Asaji T, Tanizaki Y et al. Cerebral thrombosis at altitudes: Its pathogenesis and problems of prevention and treatment. Aviation Space Environmental Med .1986; 51: 71–76.
13. Dickinson J, Heath D, Goshney J, Williams D. Altitude related death in seven trekkers in Himalya. Thorax 1983; 38: 646–656.
14. Jha SK, Anand AC, Sharma V, et al. Stroke at high altitude: Indian experience. High Alt Med Biol 2002; 3:21–27.
15. Rana PVS, Khanna LM, Suri ML, Pratapa Rao WS. Tortuosity, looping and kinking of internal carotid artery, Neurology (India) 1979; 27: 178–182.
16. Rana PVS, Suri, Ml, Vijayan GP, Pratapa Rao VVS. Ischemic cerebrovascular accident in youngs.
17. Singh I, Chauhan IS, Mathew NT fibrinolytic activity in high altitude pulmonary edema. Ind J Med Res 1969.57: 210–217.
18. Singh I, Pulmonary hypertension in new arrivals at high altitude. Proceedings of World Health Organization Meeting on Primary Pulmonary H, October l973, Geneva, WHO, 1974.
19. Severinghaus JW, Hypothetical role of angiogenesis, osmotic swelling, and ischemia at high altitude. J Appl Physiol 1995,: 79: 375–379.
20. Venkataraman S, Mohapatro AK, Rana PVS. Chronic raised intracranial tension at high altitude. Paper read in 42nd Annual Conference of Neurological Society of India 1993.
21. Muller PJ, Deck JHN. Intraocular and optic nerve sheath hemorrhages in cases of sudden intracranial tension. J Neurosurgery 1973, 41: 161–164.
22. Wiedman M and Tabin GC. High altitude retinopathy and altitude illness Ophthalmology 1999; 106: 1924–1927.
23. Foley J. Benign form of intracranial hypertension- "toxic" and "ottic" hydrocephalus. Brain 1955; 78: 1–41.
24. Rahdakrishnan K (Ed). Pseudotumour cerebri and intracranial hypertension. In Reviews in Indian Neurology 2004 SB Press (P) Ltd Trivandrum, 113–142.
25. Sylaja PN. Moosa NVA, Radhakrishnan K, Sarma PS et al. differential diagnosis of patients with intracranial sinus venous thrombosis related isolated intracranial hypertension with those with idiopathic intracranial hypertension. J Neurol Sci 2003; 215: 9–12.
26. Weisberg LA. Computed tomography in benign intracranial hypertension. Neurology 1985; 35: 1075–1078.
27. Brodsky MC, Vaphiades M. Magnetic resonance imaging in pseudotumour cerebri. Ophthalmology 1998; 105: 1686–1693.
28. Farb RI, Vanek I, Scott JN et al. Idiopathic intracranial hypertension. The prevalence and morphology of sinovenous stenosis. Neurology 2003; 60: 1418–1424.
29. Orefice G, Celentano L, Scaglione M, et al. Radioisotopic cisternography in benign intracranial hypertension of young obese women : a seven.
30. Karahalios DG, Rikate HL, Khayata MH, et al. Elevated intracranial pressure as an universal mechanism in pseudotumour cerebri of varying etiology. Neurology 1996; 46: 198–202.
31. Malm J, Kristensen B, Markgren R Ekstedt J. CSF hydrodynamics in idiopathic intracranial hypertension: a long term study. Neurology 1992; 42: 851–858.
32. King JO, Mitchell PJ, Thomson KR, Tress BM. Manometry combined with cervical puncture in idiopathic intracranial hypertension. Neurology 2002; 58: 26–30.
33. Danny Brown D, Adam RD, Brenner C, Deherty MM. Pathology and injury to nerve induced by cold. J Neuropath Expt Neurol 1945; 4: 305–310.
34. Seigmund H. Neurological manifestation of frostbite. Ind J Med res l967; 67: 292–299.
35. Thomas PK, Holdroff B; Neuropathy due to physical agents. In: Dyck PJ, PK Thomas, Griffin JW, Poduslo, JF (eds) Peripheral Neuropathy. Saunders, Philadelphia, Third Edition, Vol II, Chapter 52, pp 990–1013.
36. Francis, TJR. Non freezing cold injuries: a historical review. J R Nav Med Serv 1984; 70: 134. Quoted from Thomas PK, Holdroff B. Neuropathy due to physical agents. In: PJ Dyck, PK Thomas, Griffin JW, Poduslo, JF (eds) Peripheral Neuropathy. Saunders, Philadelphia, Third Edition, Vol II, Chapter 52, pp 990–1013.
37. Suri ML, Vijayan GR Pant HC et al. Neurological manifestations of frost bite. Ind J Med Res 292–298.
38. Ervasti E. Frost bites of extremities and their sequelae. A clinical study. Acta Chir Scand (Suppl) 1962: 299: 300–306.
39. Blair JR. Sequelae to cold injury in 100 patients. Follow up of 4 years after occurrence of cold injury. JAMA 1957: 163: 1203–1208.
40. Dash MS. A study of peripheral nerve in cold injury. Proc Int Union Physiol Soc 1974; 11:407–418.
41. Kellgren JH, McGowan AJ, Hughes ESR. On deep hyperalgesia and pain Clin Sci 1948; 7: 13–17.
42. Kunesch E, Schmidy R, Nordin M et al. Peripheral nerve correlates of cutaneous anaesthesia induced by skin cooling in man. Acta Physiol Scand 1987; 129–35.
43. Ricart de Mesones A, Turan S J, Misiego M et al. Neuropathic pain and dysesthesia of the feet after Himalayan expeditions. High Alt Med Biol. 2002 3: 395–399.
44. Thomas PK, King RH, Feng FS et al. Neurological manifestations in chronic mountain sickness: the burning hand burning feet syndrome. J Neurol Neurosurg Psychiatry 2000; 69: 447–452.
(c) Annals of Neurosciences.All Rights Reserved